
Abstract
The complement inhibitor CD55/DAF is expressed on many cell types. Dysregulation of CD55 expression is associated with increased disease severity in influenza A infection and vascular complications in pathologies that involve excessive activation of the complement system. A luciferase reporter system was used to functionally analyze the single nucleotide polymorphism rs2564978 in the U937 human promonocytic cell line. The polymorphism is in the promoter of the CD55 gene, and its minor allele T is associated with a severe course of influenza A(H1N1)pdm09. A decreased activity of the CD55 promoter carrying the minor rs2564978(T) allele was observed in activated U937 cells, which provide a cell model of human macrophages. Using bioinformatics resources, PU.1 was identified as a potential transcription factor that may bind to the CD55 promoter at the rs2564978 site in an allele-specific manner. The involvement of PU.1 in modulating CD55 promoter activity was verified by a PU.1 genetic knockdown with small interfering RNAs under specific monocyte activation conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The membrane protein CD55, which is also known as the decay-accelerating factor (DAF), acts as an inhibitor of the complement system and is broadly expressed on immune, stromal, epithelial, and endothelial cells [1]. The CD55 gene is in chromosome 1 and has 23 characterized transcripts, of which ten are protein coding according to Ensembl [2]. Based on the FANTOM5 hg38 human promoterome (https://fantom.gsc.riken.jp/zenbu/), a main transcription start site is in the 5′-untranslated region (5′-UTR) of exon 1, which is common for all protein-coding isoforms [3]. CD55 is found in a soluble form, but is most abundant in a membrane form, which contains a C-terminal glycosylphosphatidylinositol anchor [4]. CD55 protects host cells from the complement system by destabilizing convertases C3 and C5 [5]. The complement system is part of innate immunity and consists of a cascade of proteolytic interactions, which lead to direct elimination of a pathogen or an infected cell and recruitment of immune cells involved in the inflammatory response [6]. In addition, CD55 expressed on monocytes can play a role in the adaptive immune response by interacting with the adhesion molecule CD97 on the T-cell surface and thus inhibiting T-cell activity [7].
Congenital defects in CD55 expression are associated with autoimmune disorders [8, 9], CHAPLE (complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy) [10], and paroxysmal nocturnal hemoglobinuria [11]. A decrease in CD55 expression and consequent activation of the complement system are associated with microvascular complications in type 2 diabetes mellitus [12, 13]. Patients with ischemic heart disease have displayed lower CD55 expression on the monocyte surface as compared with healthy individuals [14]. An increase in CD55 expression is observed in many oncology diseases (colorectal, gastric, and ovarian cancers, leukemia, etc.), leading to lower complement-dependent anticancer cytotoxicity [1]. СD55 acts as a cell receptor of certain enteroviruses and may thus facilitate the progression of virus infections [15]. Human immunodeficiency virus type 1 (HIV-1), human T-lymphotropic virus type 1 (HTLV-1), human cytomegalovirus (HCMV) and certain other viruses can include CD55 in their virions to evade the complement system [16]. It has been shown that CD55 is involved in malaria plasmodium penetration into erythrocytes, but probably performs a protective function in cerebral malaria, in which the complement system plays an important role [17]. Thus, both excessive and insufficient activation of the complement system may affect the disease severity and mortality rate in infections because complex interactions occur between complement components and complement regulators, pathogens, and effector cells. For example, the matrix protein M1 blocks the interaction between IgG and C1qA and thus inhibits the classical complement activation pathway in influenza A infection [18], however, it is also known that dysregulation of CD55 increases the disease severity in influenza A infection because the complement system is hyperactivated and host cells are damaged [19]. A similar situation is observed in SARS-CoV-2 infection; i.e., excessive activation of the complement system is accompanied by a compensatory increase in CD55 expressed on monocytes [20].
The promoter region of CD55 harbors the single nucleotide polymorphism (SNP) rs2564978(C>T), whose minor allele T is associated with a severe course of influenza A(H1N1)pdm09 [21–23]. The CD55 mRNA level in monocytes of infected patients with the genotype rs2564978(TT) is known to be far lower than in carriers of the major allele C [21]. Wherein, the rs2564978 allele C has determined a lower activity of the CD55 promoter in lung cancer cells in reporter assays and has been associated with higher risk of non-small cell lung cancer, esophageal cancer, and enteroviral vesicular stomatitis in population studies [24–26].
In this work, we demonstrated a rs2564978-dependent regulation of CD55 promoter activity in a cell model of human macrophages and identified a potential transcription factor (TF) that may bind to the rs2564978 region in an allele-specific manner.
EXPERIMENTAL
Bioinformatics demarcation of the promoter region and a search for TF capable of allele-specific binding to the SNP region. The position of the CD55 promoter region was refined using data from ENCODE [31]. Plasmids were isolated using a Plasmid Midiprep kit (Evrogen, Russia) and verified by Sanger sequencing (Genom Collective Use Center, Russia).
Cell culture, transfection, and luciferase assay. U937 monocytes were cultured in the RPMI 1640 medium (PanEco, Russia) supplemented with 10% fetal bovine serum (FBS) (Corning, United States), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 µg/mL streptomycin (PanEco), 1% nonessential amino acids, and 10 mM HEPES (Gibco, United States). Cells were activated by supplementing the cultures with 1 µg/mL phorbol-12-myristate 13-acetate (PMA) (Sigma-Aldrich, United States) and/or 100 ng/mL lipopolysaccharide (LPS) (Escherichia coli O111:B4, L2630, Sigma-Aldrich) 24 h before transfection with plasmids; phosphate-buffered saline (PBS) was added to activation control samples. Cells were transfected via electroporation using a Neon transfection system (Thermo Scientific, United States). A sample containing 2.5 × 106 cells was combined with 5 µg of test plasmids and 0.5 µg of the control vector pRL-CMV (Promega), which expressed Renilla luciferase under a potent CMV promoter. Cell electroporation was performed using a single pulse with a voltage of 1400 V and a duration of 30 ms. Cells were lysed with a Dual-Luciferase Reporter Assay system (Promega) 24 h after transfection, and the signals produced by firefly and Renilla luciferases were measured using a 20/20n luminometer (Turner BioSystems, United States) according to the manufacturer’s protocol.
Genetic knockdown of SPI1 (PU.1) and gene expression assays. Expression of the SPI1 (PU.1) gene in U937 cells was suppressed via RNA interference with small interfering RNAs (siRNAs) complementary to coding gene regions. Pairs of siRNAs to perform a SPI1 knockdown and control scrambled siRNAs (Table 1) were synthesized by DNA-Syntez LLC (Russia) on the basis of published sequences [32]. To obtain a siRNA duplex, equimolar amounts of sense and antisense oligonucleotides were combined in a buffer (10 mM Tris, 20 mM NaCl, pH 8.0), heated at 98°C, and slowly chilled at room temperature. On day 1, U937 were electroporated with siRNA duplexes, which were used at 500 pmol per 2.5 × 106 cells. Then the cells were activated with PMA + LPS 24 h after electroporation. On day 3, the cells were electroporated using the test constructs and 300 pmol of the same siRNA duplex. On day 4, luciferase activities were measured by the dual luciferase assay and part of the cells was taken for total RNA isolation and gene expression measurements. Total RNA was isolated using the ExtractRNA reagent (Evrogen, Russia) according to the manufacturer’s protocol. Reverse transcription was carried out with oligo(dT)18 primers and a MMLV RT kit (Evrogen) according to the manufacturer’s protocol. The resulting cDNA concentration was measured using a Real-time CFX96 Touch instrument (Bio-Rad, United States) with a qPCRmix-HS SYBR ready mix (Evrogen) and specific primers (Table 1) as recommended by the manufacturer.
Statistical analyses were carried out using GraphPad Prism (v. 9.0.0. for Windows, GraphPad Software, United States; www.graphpad.com). Statistical significance was assessed by Student’s unpaired t-test. Data were collected in at least three independent experiments and presented as mean ± standard error of the mean (SEM). Differences were considered significant at P < 0.05.
RESULTS
Activity of CD55 Promoter with Minor Allele rs2564978(Т) Is Lower Than with Major Allele C in Cell Model of Human Macrophages
Human monocytes/macrophages are known to play an important role in the antivirus immune response [33]. The CD55 mRNA level in monocytes in virus-infected patients with the genotype rs2564978(TT) is substantially lower than in carriers of the allele C [21]. Based on the FANTOM5 data (hg38, https://fantom.gsc.riken.jp/zenbu/), CD14+ monocytes display the highest CD55 expression level among all immune cells. We therefore used the U937 cell line (a model of human monocytes) to study the effects of the alternative rs2564978 alleles on the CD55 promoter.
The CD55 promoter region (chr1:207320724–207321763; GRCh38/hg38), including the rs2564978 polymorphism, was chosen based on the epigenetic marks of active chromatin in CD14+ monocytes (high levels of the H3K4me3 and H3K27ac histone modifications, DNase I hypersensitivity sites, and cluster of TF binding sites) and published data [21] (Fig. 1). The genome fragment chosen as the promoter for further analysis was approximately 1 kb in length and coded for the total CD55 5'-UTR.

Scheme of the rs2564978 position in the CD55 promoter as visualized using the UCSC Genome Browser (GRCh38/hg38). The CD55 promoter region is shaded blue. The position of the rs2564978 SNP is shown with a red vertical line. Histograms demonstrate the positions of histone modifications associated with active regulatory genome regions (H3K4 mono- and trimethylation and H3K27 acetylation; Roadmap) in CD14+ monocytes. Bars show the clusters of DNase I-hypersensitive sites in CD14+ monocytes and TF binding sites (ChIP-seq ENCODE). ChromHMM characterizes chromatin activity in several CD14+ monocyte subpopulations (according to Roadmap); promoter-like and enhancer-like regions are shown red and orange, respectively.
The CD55 promoter with the alternative variants of the rs2564978 polymorphism was cloned in pGL3-basic (Fig. 2a), and the resulting reporter constructs were used to transfect U937 cells without activation (PBS was added) and U937 cells stimulated with PMA or PMA + LPS. Activation with PMA is known to stimulate differentiation of U937 monocytes into macrophate-like cells with the M0-like phenotype, while cell stimulation with both PMA and LPS polarizes the cells to a M1-like phenotype [34, 35]. Reporter gene expression under the control of the CD55 promoter in U937 cells increased substantially after cell activation (Fig. 2b), and expression of the endogenous CD55 mRNA also tended to increase (Fig. 2c). Nonstimulated monocytes did not display a significant difference in activity between the promoters carrying the alternative rs2564978 alleles. In stimulated cells, the promoter carrying the minor allele rs2564978(T) was less active than the promoter carrying the major allele C (Fig. 2b).

Activity of the CD55 promoter is lower in the presence of the minor rs2564978(Т) allele in a cell model of macrophages. (a) Scheme of the luciferase reporter construct pGL3-basic containing the CD55 promoter with the alternative rs2564978 alleles. (b) Stimulation of U937 monocytes with PMA or PMA + LPS increases the allele-dependent difference in CD55 promoter activity. Expression of reporter firefly luciferase was normalized to that of Renilla luciferase (internal control). (c) Relative CD55 mRNA expression in nonactivated U937 cells (PBS) and U937 cells stimulated with PMA or PMA + LPS. Results of three replicate experiments are shown as mean ± SEM. * A difference between the rs2564978(С) and rs2564978(Т) variants was significant at P < 0.05; (###) a difference between nonstimulated (PBS) and activated cells was significant at P < 0.0001 (Student’s unpaired t-test).
Transcription Factor PU.1 (SPI1) Is Involved in Allele-Dependent Effect of rs2564978 Polymorphism on CD55 Promoter Activity in Macrophage Cell Model
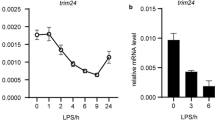
A difference in activity between regulatory elements carrying alternative SNP variants may be determined by the allele-specific binding of a particular TF in the SNP region [36, 37]. Potential TFs that bind to the rs2564978 region in an allele-specific manner were sought using the ADASTRA bioinformatics resource, which is based on a meta-analysis of ChIP-seq data [30]. According to ADASTRA, the TF PU.1 is predicted to more efficiently bind to the CD55 promoter region containing the major rs2564978(С) allele than to the region with the minor T variant in CD14+ monocytes (Fig. 3a). PU.1 is encoded by the SPI1 gene and acts as a main regulator of myeloid cell differentiation [38]. We studied how the SPI1 transcription level changes with activation of U937 monocytes. The highest expression of the endogenous SPI1 mRNA was observed upon polarization of U937 monocytes to macrophage-like cells (Fig. 3b). The finding agreed with the results of the reporter analysis (Fig. 2b).

A genetic knockdown of PU.1 (SPI1) decreases the activity of the CD55 promoter containing the major rs2564978(C) allele in activated U937 monocytes. (a) A sequence logo of the PU.1 (SPI1) binding site from the database HOCOMOCO v. 11 and the aligned sequences of the CD55 promoter with different rs2564978 alleles (shaded). P-values of the respective motifs are shown at the top of the sequences to characterize the predicted PU.1 binding specificity for the alternative alleles. (b) Relative expression of the SPI1 mRNA in nonactivated U937 cells (PBS) and U937 cells stimulated with PMA or PMA + LPS. A difference in SPI1 mRNA expression between U937 cells stimulated with PMA + LPS and nonstimulated U937 cells was significant at P < 0.05. (c) Relative SPI1 mRNA expression in U937 cells knocked down in SPI1. * P < 0.05 (Student’s unpaired t-test). (d) Relative activity of reporter luciferase expressed under the CD55 promoter with different rs2564978 variants in activated U937 cells with a SPI1 knockdown. Normalization was performed to Renilla luciferase activity as an internal control. Nonmodified cells (control) and cells transfected with nonspecific siRNA (siRNA-scr) were used as controls. (*) A difference between the rs2564978(С) and rs2564978(Т) variants was significant at P < 0.05 (Student’s unpaired t-test). (#) Differences in activity of the rs2564978(С) promoter activity between cells knocked down in SPI1 and control or siRNA-scr cells were significant at P < 0.05. (e) Relative CD55 mRNA expression in cells knocked down in SPI1. Results from three replicate experiments are shown as mean ± SEM.
To verify the PU.1 involvement in the allele-dependent regulation of CD55 promoter activity, a SPI1 genetic knockdown was achieved in activated U937 cells with siRNAs. The efficiency of SPI1 transcriptional suppression was higher than 90% (Fig. 3c). It is of interest that inhibition of PU.1 expression in activated monocytes eliminated the difference between the CD55 promoter variants carrying the alternative alleles of rs2564978 (Fig. 3d), but expression of the endogenous CD55 mRNA did not decrease significantly (Fig. 3e). The findings indicate that the TF PU.1 is potentially involved in the allele-dependent effect of rs2564978 on CD55 promoter activity, binding less efficiently to the region with rs2564978(Т).
DISCUSSION
Many associations of SNPs with various disorders have been characterized to date in genome-wide association studies (GWASs), and approximately 95% of clinically significant SNPs are in noncoding regulatory regions of the genome [39]. This distribution is possibly related to changes in the characteristics of regulatory regions around SNPs with consequent effects on gene expression; i.e., SNPs act as expression quantitative trait loci (eQTLs) of pathogenetically important genes [40]. In this work, we used the genetic constructs containing the CD55 promoter with the 21‑bp insertion rs28371582(ins), which is 68 bp proximal of rs2564978. According to LDpair Tool [41], rs2564978 and rs28371582 (also known as rs3841376 or rs150046210) are in linkage disequilibrium (LD) in the global population. The genotype rs2564978(С) occurs predominantly with rs28371582(ins), while the genotype rs2564978(Т), with the deletion variant rs28371582(del), except in the African population. In addition, the combinations rs2564978(С)/ rs28371582(ins) and rs2564978(Т)/rs28371582(del) are common in the Asian population. The genotype rs2564978(Т)/rs28371582(del) has been associated with lower activity of the CD55 promoter as compared with the genotype rs2564978(C)/rs28371582(ins) in bronchial epithelial cells (BEAS-2B) [21]. We studied the direct effect of the rs2564978 polymorphism on the activity of the CD55 promoter in activated monocytes. The results indicate that CD55 promoter activity is lower in the presence of the minor rs2564978(T) allele, which is associated with a severe course of influenza А(H1N1)pdm09, and that a decrease in activity is possibly due to a distorted binding of PU.1 to the region containing rs2564978.
Seasonal respiratory infections are caused by the influenza virus mostly because of its fast airborne transmission and the development of secondary bacterial infections and superinfections [19, 43]. Cells infected with the influenza virus expose the viral proteins hemagglutinin and neuraminidase on their surfaces. The latter acts as a potent sialidase and cleaves the sialic acid moiety of CD55. Inhibition of C3 convertase is distorted as a result, and the complement system is hyperactivated. Moreover, the presence of hemagglutinin on the cell membrane and the recognition of desialilated surface proteins by pattern recognition receptors may lead to enhanced activation of the immune response and excessive inflammation, thereby facilitating damage to tissues and determining a poorer outcome [6]. Thus, a decrease in complement inhibitor CD55 on the macrophage surface possibly explains the association of rs2564978(Т) with a severe course of influenza A infection, and the TF PU.1 may act as a molecular mediator of the process (Fig. 4).

Hypothetical scheme of the allele-dependent effect of rs2564978 on the development of pathological complications due to hyperactivation of the complement system (created with BioRender.com).
It is of interest that the major allele rs2564978(C) is associated with higher susceptibility to enteroviral vesicular stomatitis and higher risk of non-small cell lung cancer and esophageal cancer [25, 26]. As reporter assays have shown, activity of the CD55 promoter containing rs2564978(T) is higher than that of the promoter with rs2564978(C) in cell models of lung cancer (A549, NCI-H2030, and NCI-H23) [24], while no significant difference in activity has been observed between the CD55 promoter variants carrying the alternative rs2564978 alleles in normal lung epithelial cells (BEAS-2B) [21]. The data suggest tissue specificity for the mechanisms that sustain the effect of rs2564978 on CD55 expression. The TF PU.1 possibly acts as a main regulator in myeloid cells according to our findings, while another TF may be responsible for the allele-dependent effect of rs2564978 on CD55 promoter activity in epithelial cells, where the effect of the polymorphism is opposite. For example, the multifunctional TF CTCF more efficiently binds around rs2564978(T) in the CD55 promoter in the esophageal epithelium according to ADASTRA. Hence CTCF may determine the allele-dependent effect in cells of this type because PU.1 is expressed to a low level in epithelial cells (according to FANTOM5 hg38 human promoterome).
It should also be noted that hyperactivation of the complement system promotes damage to vascular endothelial cells, which is thought to determine a more severe course in virus infections (flu and COVID-19) [44] and cardiovascular complications in other pathologies. For example, a low level of CD55 expression has been observed in patients with type 2 diabetes mellitus, nephropathy, and retinopathy [46]. We assume that a rs2564978(T)-dependent decrease in CD55 expression on monocytes and macrophages may contribute to the genetic predisposition to excessive complement activation, which causes a broad range of microvascular complications.
ABBREVIATIONS
DAF, decay-accelerating factor; SNP, single nucleotide polymorphism; ChIP-seq, chromatin immunoprecipitation sequencing; LPS, lipopolysaccharide; PMA, phorbol-12-myristate 13-acetate; PBS, phosphate-buffered saline; siRNA, small interfering RNA; 5′-UTR, 5′-untranslated region; TF, transcription factor.
REFERENCES
Dho S.H., Lim J.C., Kim L.K. 2018. Beyond the role of CD55 as a complement component. Immune Networks. 18 (1), e11.
Cunningham F., Allen J.E., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Austine-Orimoloye O., Azov A.G., Barnes I., Bennett R., Berry A., Bhai J., Bignell A., Billis K., Boddu S., Brooks L., Charkhchi M., Cummins C., Da Rin Fioretto L., Davidson C., Dodiya K., Donaldson S., El Houdaigui B., El Naboulsi T., Fatima R., Giron C.G., Genez T., Martinez J.G., Guijarro-Clarke C., Gymer A., Hardy M., Hollis Z., Hourlier T., Hunt T., Juettemann T., Kaikala V., Kay M., Lavidas I., Le T., Lemos D., Marugán J.C., Mohanan S., Mushtaq A., Naven M., Ogeh D.N., Parker A., Parton A., Perry M., Pilizota I., Prosovetskaia I., Sakthivel M.P., Salam A.I.A., Schmitt B.M., Schuilenburg H., Sheppard D., Perez-Silva J.G., Stark W., Steed E., Sutinen K., Sukumaran R., Sumathipala D., Suner M.M., Szpak M., Thormann A., Tricomi F.F., Urbina-Gómez D., Veidenberg A., Walsh T.A., Walts B., Willhoft N., Winterbottom A., Wass E., Chakiachvili M., Flint B., Frankish A., Giorgetti S., Haggerty L., Hunt S.E., Iisley G.R., Loveland J.E., Martin F.J., Moore B., Mudge J.M., Muffato M., Perry E., Ruffier M., Tate J., Thybert D., Trevanion S.J., Dyer S., Harrison P.W., Howe K.L., Yates A.D., Zerbino D.R., Flicek P. 2022. Ensembl 2022. Nucl. Acids Res. 50 (D1), D988.
Forrest A.R.R., Kawaji H., Rehli M., Baillie J.K., De Hoon M.J.L., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., Itoh M., Andersson R., Mungall C.J., Meehan T.F., Schmeier S., Bertin N., Jørgensen M., Dimont E., Arner E., Schmidl C., Schaefer U., Medvedeva Y.A., Plessy C., Vitezic M., Severin J., Semple C.A., Ishizu Y., Young R.S., Francescatto M., Altschuler I.A., Albanese D., Altschule G.M., Arakawa T., Archer J.A.C., Arner P., Babina M., Rennie S., Balwierz P.J., Beckhouse A.G., Pradhan-Bhatt S., Blake J.A., Blumenthal A., Bodega B., Bonetti A., Briggs J., Brombacher F., Burroughs A.M., Califano A., Cannistraci C.V., Carbajo D., Chen Y., Chierici M., Ciani Y., Clevers H.C., Dalla E., Davis C.A., Detmar M., Diehl A.D., Dohi T., Drabløs F., Edge A.S.B., Edinger M., Ekwall K., Endoh M., Enomoto H., Fagiolini M., Fairbairn L., Fang H., Farach-Carson M.C., Faulkner G.J., Favorov A.V., Fisher M.E., Frith M.C., Fujita R., Fukuda S., Furlanello C., Furuno M., Furusawa J.I., Geijtenbeek T.B., Gibson A.P., Gingeras T., Goldowitz D., Gough J., Guhl S., Guler R., Gustincich S., Ha T.J., Hamaguchi M., Hara M., Harbers M., Harshbarger J., Hasegawa A., Hasegawa Y., Hashimoto T., Herlyn M., Hitchens K.J., Sui S.J.H., Hofmann O.M., Hoof I., Hori F., Huminiecki L., Iida K., Ikawa T., Jankovic B.R., Jia H., Joshi A., Jurman G., Kaczkowski B., Kai C., Kaida K., Kaiho A., Kajiyama K., Kanamori-Katayama M., Kasianov A.S., Kasukawa T., Katayama S., Kato S., Kawaguchi S., Kawamoto H., Kawamura Y.I., Kawashima T., Kempfle J.S., Kenna T.J., Kere J., Khachigian L.M., Kitamura T., Klinken S.P., Knox A.J., Kojima M., Kojima S., Kondo N., Koseki H., Koyasu S., Krampitz S., Kubosaki A., Kwon A.T., Laros J.F.J., Lee W., Lennartsson A., Li K., Lilje B., Lipovich L., Mackay-sim A., Manabe R.I., Mar J.C., Marchand B., Mathelier A., Mejhert N., Meynert A., Mizuno Y., De Morais D.A.L., Morikawa H., Morimoto M., Moro K., Motakis E., Motohashi H., Mummery C.L., Murata M., Nagao-Sato S., Nakachi Y., Nakahara F., Nakamura T., Nakamura Y., Nakazato K., Van Nimwegen E., Ninomiya N., Nishiyori H., Noma S., Nozaki T., Ogishima S., Ohkura N., Ohmiya H., Ohno H., Ohshima M., Okada-Hatakeyama M., Okazaki Y., Orlando V., Ovchinnikov D.A., Pain A., Passier R., Patrikakis M., Persson H., Piazza S., Prendergast J.G.D., Rackham O.J.L., Ramilowski J.A., Rashid M., Ravasi T., Rizzu P., Roncador M., Roy S., Rye M.B., Saijyo E., Sajantila A., Saka A., Sakaguchi S., Sakai M., Sato H., Satoh H., Savvi S., Saxena A., Schneider C., Schultes E.A., Schulze-Tanzil G.G., Schwegmann A., Sengstag T., Sheng G., Shimoji H., Shimoni Y., Shin J.W., Simon C., Sugiyama D., Sugiyama T., Suzuki M., Suzuki N., Swoboda R.K., ’T Hoen P.A.C., Tagami M., Tagami N.T., Takai J., Tanaka H., Tatsukawa H., Tatum Z., Thompson M., Toyoda H., Toyoda T., Valen E., Van De Wetering M., Van Den Berg L.M., Verardo R., Vijayan D., Vorontsov I.E., Wasserman W.W., Watanabe S., Wells C.A., Winteringham L.N., Wolvetang E., Wood E.J., Yamaguchi Y., Yamamoto M., Yoneda M., Yonekura Y., Yoshida S., Zabierowski S.E., Zhang P.G., Zhao X., Zucchelli S., Summers K.M., Suzuki H., Daub C.O., Kawai J., Heutink P., Hide W., Freeman T.C., Lenhard B., Bajic L.V.B., Taylor M.S., Makeev V.J., Sandelin A., Hume D.A., Carninci P., Hayashizaki Y. 2014. A promoter-level mammalian expression atlas. Nature. 507 (7493), 462–470.
Vainer E.D., Meir K., Furman M., Semenenko I., Konikoff F., Vainer G.W. 2013. Characterization of novel CD55 isoforms expression in normal and neoplastic tissues. Tissue Antigens. 82 (1), 26–34.
Geller A., Yan J. 2019. The role of membrane bound complement regulatory proteins in tumor development and cancer immunotherapy. Front. Immunol. 10, 1074.
Santos N.B., Vaz Da Silva Z.E., Gomes C., Reis C.A., Amorim M.J. 2021. Complement decay-accelerating factor is a modulator of influenza A virus lung immunopathology. PLoS Pathog. 17 (7), e1009381.
Abbott R.J.M., Spendlove I., Roversi P., Fitzgibbon H., Knott V., Teriete P., McDonnell J.M., Handford P.A., Lea S.M. 2007. Structural and functional characterization of a novel T cell receptor co-regulatory protein complex, CD97-CD55. J. Biol. Chem. 282 (30), 22023–22032.
Hamann J., Wishaupt J.O., Van Lier R.A.W., Smeets T.J.M., Breedveld F.C., Tak P.P. 1999. Expression of the activation antigen CD97 and its ligand CD55 in rheumatoid synovial tissue. Arthritis Rheum. 42 (4), 650‒658.
Miwa T., Maldonado M.A., Zhou L., Yamada K., Gilkeson G.S., Eisenberg R.A., Song W.C. 2007. Decay-accelerating factor ameliorates systemic autoimmune disease in MRL/Ipr mice via both complement-dependent and -independent mechanisms. Am. J. Pathol. 170 (4), 1258‒1266.
Ozen A., Comrie W.A., Ardy R.C., Domínguez Conde C., Dalgic B., Beser Ö.F., Morawski A.R., Karakoc-Aydiner E., Tutar E., Baris S., Ozcay F., Serwas N.K., Zhang Y., Matthews H.F., Pittaluga S., Folio L.R., Unlusoy Aksu A., McElwee J.J., Krolo A., Kiykim A., Baris Z., Gulsan M., Ogulur I., Snapper S.B., Houwen R.H.J., Leavis H.L., Ertem D., Kain R., Sari S., Erkan T., Su H.C., Boztug K., Lenardo M.J. 2017. CD55 deficiency, early-onset protein-losing enteropathy, and thrombosis. N. Engl. J. Med. 377 (1), 52‒61.
Brodsky R.A. 2008. Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Rev. 22 (2), 65‒74.
Ma X.W., Chang Z.W., Qin M.Z., Sun Y., Huang H.L., He Y. 2009. Decreased expression of complement regulatory proteins, CD55 and CD59, on peripheral blood leucocytes in patients with type 2 diabetes and macrovascular diseases. Chin. Med. J. 122 (18), 2123‒2128.
Zhang J., Gerhardinger C., Lorenzi M. 2002. Early complement activation and decreased levels of glycosylphosphatidylinositol-anchored complement inhibitors in human and experimental diabetic retinopathy. Diabetes. 51 (12), 3499‒3504.
Mishra N., Mohata M., Narang R., Lakshmy R., Hazarika A., Pandey R.M., Das N., Luthra K. 2019. Altered expression of complement regulatory proteins CD35, CD46, CD55, and CD59 on leukocyte subsets in individuals suffering from coronary artery disease. Front. Immunol. 10, 2072.
Bergelson J.M., Chan M., Solomon K.R., St. John N.F., Lin H., Finberg R.W. 1994. Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc. Natl. Acad. Sci. U. S. A. 91 (13), 6245‒6248.
Stoermer K.A., Morrison T.E. 2011. Complement and viral pathogenesis. Virol. J. 411 (2), 362–373.
Biryukov S., Stoute J.A. 2014. Complement activation in malaria: Friend or foe? Trends Mol. Med. 20 (5), 293‒301.
Zhang J., Li G., Liu X., Wang Z., Liu W., Ye X. 2009. Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J. Gen. Virol. 90 (Pt. 11), 2751–2758.
Nogales A., Dediego M.L. 2019. Host single nucleotide polymorphisms modulating influenza a virus disease in humans. Pathogens. 8 (4), 168.
Lage S.L., Rocco J.M., Laidlaw E., Rupert A., Galindo F., Kellogg A., Kumar P., Poon R., Wortmann G.W., Lisco A., Manion M., Sereti I. 2022. Activation of complement components on circulating blood monocytes from COVID-19 patients. Front. Immunol. 13, 815833.
Zhou J., To K.K.W., Dong H., Cheng Z.S., Lau C.C.Y., Poon V.K.M., Fan Y.H., Song Y.Q., Tse H., Chan K.H., Zheng B.J., Zhao G.P., Yuen K.Y. 2012. A functional variation in CD55 increases the severity of 2009 pandemic H1N1 influenza a virus infection. J. Infect. Dis. 206 (4), 495‒503.
Chatzopoulou F., Gioula G., Kioumis I., Chatzidimitriou D., Exindari M. 2019. Identification of complement-related host genetic risk factors associated with influenza A(H1N1)pdm09 outcome: Challenges ahead. Med. Microbiol. Immunol. 208 (5), 631‒640.
Lee N., Cao B., Ke C., Lu H., Hu Y., Tam C.H.T., Ma R.C.W., Guan D., Zhu Z., Li H., Lin M., Wong R.Y.K., Yung I.M.H., Hung T.-N., Kwok K., Horby P., Hui D.S.C., Chan M.C.W., Chan P.K.S. 2017. Single-nucleotide polymorphisms of IFITM3, TLR3, CD55, and TLR4 and risk for severe outcomes in patients with influenza A (H7N9) and (H1N1) pdm09 in China: A multicentre cohort study. Lancet. 390, S1.
Zhang Y., Zhang Z., Cao L., Lin J., Yang Z., Zhang X. 2017. A common CD55 rs2564978 variant is associated with the susceptibility of non-small cell lung cancer. Oncotarget. 8 (4), 6216‒6221.
Wu H.J., Gao H., **e Y.N., Zhang Y.Y., Yang Z.B., Zhang X.M. 2018. A promoter polymorphism of CD55 effect on the risk of esophageal cancer. Zhonghua yu fang yi xue za zhi [Chin. J. Prev. Med.]. 52 (8), 822–826.
Li M., Li Y.P., Deng H.L., Wang M.Q., Wang W.J., Wang J., Wu F.P., Dang S.S. 2020. Association of gene polymorphisms of CD55 with susceptibility to and severity of hand, foot, and mouth disease caused by enterovirus 71 in the Han Chinese population. J. Med. Virol. 92 (12), 3119‒3124.
Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., Khatun J., Lajoie B.R., Landt S.G., Lee B.K., Pauli F., Rosenbloom K.R., Sabo P., Safi A., Sanyal A., Shoresh N., Simon J.M., Song L., Trinklein N.D., Altshuler R.C., Birney E., Brown J.B., Cheng C., Djebali S., Dong X., Ernst J., Furey T.S., Gerstein M., Giardine B., Greven M., Hardison R.C., Harris R.S., Herrero J., Hoffman M.M., Iyer S., Kellis M., Kheradpour P., Lassmann T., Li Q., Lin X., Marinov G.K., Merkel A., Mortazavi A., Parker S.C.J., Reddy T.E., Rozowsky J., Schlesinger F., Thurman R.E., Wang J., Ward L.D., Whitfield T.W., Wilder S.P., Wu W., ** H.S., Yip K.Y., Zhuang J., Bernstein B.E., Green E.D., Gunter C., Snyder M., Pazin M.J., Lowdon R.F., Dillon L.A.L., Adams L.B., Kelly C.J., Zhang J., Wexler J.R., Good P.J., Feingold E.A., Crawford G.E., Dekker J., Elnitski L., Farnham P.J., Giddings M.C., Gingeras T.R., Guigó R., Hubbard T.J., Kent W.J., Lieb J.D., Margulies E.H., Myers R.M., Stamatoyannopoulos J.A., Tenenbaum S.A., Weng Z., White K.P., Wold B., Yu Y., Wrobel J., Risk B.A., Gunawardena H.P., Kuiper H.C., Maier C.W., **e L., Chen X., Mikkelsen T.S., Gillespie S., Goren A., Ram O., Zhang X., Wang L., Issner R., Coyne M.J., Durham T., Ku M., Truong T., Eaton M.L., Dobin A., Tanzer A., Lagarde J., Lin W., Xue C., Williams B.A., Zaleski C., Röder M., Kokocinski F., Abdelhamid R.F., Alioto T., Antoshechkin I., Baer M.T., Batut P., Bell I., Bell K., Chakrabortty S., Chrast J., Curado J., Derrien T., Drenkow J., Dumais E., Dumais J., Duttagupta R., Fastuca M., Fejes-Toth K., Ferreira P., Foissac S., Fullwood M.J., Gao H., Gonzalez D., Gordon A., Howald C., Jha S., Johnson R., Kapranov P., King B., Kingswood C., Li G., Luo O.J., Park E., Preall J.B., Presaud K., Ribeca P., Robyr D., Ruan X., Sammeth M., Sandhu K.S., Schaeffer L., See L.H., Shahab A., Skancke J., Suzuki A.M., Takahashi H., Tilgner H., Trout D., Walters N., Wang H., Hayashizaki Y., Reymond A., Antonarakis S.E., Hannon G.J., Ruan Y., Carninci P., Sloan C.A., Learned K., Malladi V.S., Wong M.C., Barber G.P., Cline M.S., Dreszer T.R., Heitner S.G., Karolchik D., Kirkup V.M., Meyer L.R., Long J.C., Maddren M., Raney B.J., Grasfeder L.L., Giresi P.G., Battenhouse A., Sheffield N.C., Showers K.A., London D., Bhinge A.A., Shestak C., Schaner M.R., Kim S.K., Zhang Z.Z., Mieczkowski P.A., Mieczkowska J.O., Liu Z., McDaniell R.M., Ni Y., Rashid N.U., Kim M.J., Adar S., Zhang Z., Wang T., Winter D., Keefe D., Iyer V.R., Zheng M., Wang P., Gertz J., Vielmetter J., Partridge E.C., Varley K.E., Gasper C., Bansal A., Pepke S., Jain P., Amrhein H., Bowling K.M., Anaya M., Cross M.K., Muratet M.A., Newberry K.M., McCue K., Nesmith A.S., Fisher-Aylor K.I., Pusey B., DeSalvo G., Parker S.L., Balasubramanian S., Davis N.S., Meadows S.K., Eggleston T., Newberry J.S., Levy S.E., Absher D.M., Wong W.H., Blow M.J., Visel A., Pennachio L.A., Petrykowska H.M., Abyzov A., Aken B., Barrell D., Barson G., Berry A., Bignell A., Boychenko V., Bussotti G., Davidson C., Despacio-Reyes G., Diekhans M., Ezkurdia I., Frankish A., Gilbert J., Gonzalez J.M., Griffiths E., Harte R., Hendrix D.A., Hunt T., Jungreis I., Kay M., Khurana E., Leng J., Lin M.F., Loveland J., Lu Z., Manthravadi D., Mariotti M., Mudge J., Mukherjee G., Notredame C., Pei B., Rodriguez J.M., Saunders G., Sboner A., Searle S., Sisu C., Snow C., Steward C., Tapanari E., Tress M.L., Van Baren M.J., Washietl S., Wilming L., Zadissa A., Zhang Z., Brent M., Haussler D., Valencia A., Addleman N., Alexander R.P., Auerbach R.K., Balasubramanian S., Bettinger K., Bhardwaj N., Boyle A.P., Cao A.R., Cayting P., Charos A., Cheng Y., Eastman C., Euskirchen G., Fleming J.D., Grubert F., Habegger L., Hariharan M., Harmanci A., Iyengar S., ** V.X., Karczewski K.J., Kasowski M., Lacroute P., Lam H., Lamarre-Vincent N., Lian J., Lindahl-Allen M., Min R., Miotto B., Monahan H., Moqtaderi Z., Mu X.J., O’Geen H., Ouyang Z., Patacsil D., Raha D., Ramirez L., Reed B., Shi M., Slifer T., Witt H., Wu L., Xu X., Yan K.K., Yang X., Struhl K., Weissman S.M., Penalva L.O., Karmakar S., Bhanvadia R.R., Choudhury A., Domanus M., Ma L., Moran J., Victorsen A., Auer T., Centanin L., Eichenlaub M., Gruhl F., Heermann S., Hoeckendorf B., Inoue D., Kellner T., Kirchmaier S., Mueller C., Reinhardt R., Schertel L., Schneider S., Sinn R., Wittbrodt B., Wittbrodt J., Jain G., Balasundaram G., Bates D.L., Byron R., Canfield T.K., Diegel M.J., Dunn D., Ebersol A.K., Frum T., Garg K., Gist E., Hansen R.S., Boatman L., Haugen E., Humbert R., Johnson A.K., Johnson E.M., Kutyavin T. V., Lee K., Lotakis D., Maurano M.T., Neph S.J., Neri F. V., Nguyen E.D., Qu H., Reynolds A.P., Roach V., Rynes E., Sanchez M.E., Sandstrom R.S., Shafer A.O., Stergachis A.B., Thomas S., Vernot B., Vierstra J., Vong S., Wang H., Weaver M.A., Yan Y., Zhang M., Akey J.M., Bender M., Dorschner M.O., Groudine M., MacCoss M.J., Navas P., Stamatoyannopoulos G., Beal K., Brazma A., Flicek P., Johnson N., Lukk M., Luscombe N.M., Sobral D., Vaquerizas J.M., Batzoglou S., Sidow A., Hussami N., Kyriazopoulou-Panagiotopoulou S., Libbrecht M.W., Schaub M.A., Miller W., Bickel P.J., Banfai B., Boley N.P., Huang H., Li J.J., Noble W.S., Bilmes J.A., Buske O.J., Sahu A.D., Kharchenko P. V., Park P.J., Baker D., Taylor J., Lochovsky L. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57–74.
Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J., Amin V., Whitaker J.W., Schultz M.D., Ward L.D., Sarkar A., Quon G., Sandstrom R.S., Eaton M.L., Wu Y.-C., Pfenning A.R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R.A., Shoresh N., Epstein C.B., Gjoneska E., Leung D., **e W., Hawkins R.D., Lister R., Hong C., Gascard P., Mungall A.J., Moore R., Chuah E., Tam A., Canfield T.K., Hansen R.S., Kaul R., Sabo P.J., Bansal M.S., Carles A., Dixon J.R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T.R., Neph S.J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R.C., Siebenthall K.T., Sinnott-Armstrong N.A., Stevens M., Thurman R.E., Wu J., Zhang B., Zhou X., Beaudet A.E., Boyer L.A., Jager P.L. De, Farnham P.J., Fisher S.J., Haussler D., Jones S.J.M., Li W., Marra M.A., McManus M.T., Sunyaev S., Thomson J.A., Tlsty T.D., Tsai L.-H., Wang W., Waterland R.A., Zhang M.Q., Chadwick L.H., Bernstein B.E., Costello J.F., Ecker J.R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J.A., Wang T., Kellis M. 2015. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317–330.
Ernst J., Kellis M. 2017. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 12 (12), 2478–2492.
Abramov S., Boytsov A., Bykova D., Penzar D.D., Yevshin I., Kolmykov S.K., Fridman M.V., Favorov A.V., Vorontsov I.E., Baulin E., Kolpakov F., Makeev V.J., Kulakovskiy I.V. 2021. Landscape of allele-specific transcription factor binding in the human genome. Nat. Commun. 12 (1), 2751.
Ustiugova A.S., Korneev K. V., Kuprash D. V., Afanasyeva M.A. 2019. Functional SNPs in the human autoimmunity-associated locus 17q12-21. Genes. 10 (2), 77.
Schotte R., Nagasawa M., Weijer K., Spits H., Blom B. 2004. The ETS transcription factor Spi-B is required for human plasmacytoid dendritic cell development. J. Exp. Med. 200 (11), 1503‒1509.
Roberts N.J. 2020. Diverse and unexpected roles of human monocytes/macrophages in the immune response to influenza virus. Viruses. 12 (4), 379.
Nascimento C.R., Rodrigues Fernandes N.A., Gonzalez Maldonado L.A., Rossa Junior C. 2022. Comparison of monocytic cell lines U937 and THP-1 as macrophage models for in vitro studies. Biochem. Biophys. Rep. 32, 101383.
Wang N., Liang H., & Zen K. 2014. Molecular mechanisms that influence the macrophage M1-M2 polarization balance. Front. Immunol. 5, 614.
Uvarova A.N., Stasevich E.M., Ustiugova A.S., Mitkin N.A., Zheremyan E.A., Sheetikov S.A., Zornikova K.V., Bogolyubova A.V., Rubtsov M.A., Kulakovskiy I.V., Kuprash D.V., Korneev K.V., Schwartz A.M. 2023. rs71327024 associated with COVID-19 hospitalization reduces CXCR6 promoter activity in human CD4+ T cells via disruption of c-Myb binding. Int. J. Mol. Sci. 24 (18), 13790.
Korneev K. V., Sviriaeva E.N., Mitkin N.A., Gorbacheva A.M., Uvarova A.N., Ustiugova A.S., Polanovsky O.L., Kulakovskiy I.V., Afanasyeva M.A., Schwartz A.M., Kuprash D. V. 2020. Minor C allele of the SNP rs7873784 associated with rheumatoid arthritis and type-2 diabetes mellitus binds PU.1 and enhances TLR4 expression. Biochim. Biophys. Acta, Mol. Basis Dis. 1866 (3), 165626.
Fisher R.C., Scott E.W. 1998. Role of PU.1 in hematopoiesis. Stem Cells. 16 (1), 25–37.
Orozco G., Schoenfelder S., Walker N., Eyre S., Fraser P. 2022. 3D genome organization links non-coding disease-associated variants to genes. Front. Cell Dev. Biol. 10, 995388.
Johnston A.D., Simões-Pires C.A., Thompson T. V., Suzuki M., Greally J.M. 2019. Functional genetic variants can mediate their regulatory effects through alteration of transcription factor binding. Nat. Commun. 10 (1), 3472.
Lin S.H., Thakur R., Machiela M.J. 2021. LDexpress: An online tool for integrating population-specific linkage disequilibrium patterns with tissue-specific expression data. BMC Bioinf. 22 (1), 608.
Kawai T., Takeshita S., Imoto Y., Matsumoto Y., Sakashita M., Suzuki D., Shibasaki M., Tamari M., Hirota T., Arinami T., Fujieda S., Noguchi E. 2009. Associations between decay-accelerating factor polymorphisms and allergic respiratory diseases. Clin. Exp. Allergy. 39 (10), 1508‒1514.
Sviriaeva E.N., Korneev K.V., Drutskaya M.S., Kuprash D.V. 2016. Mechanisms of changes in immune response during bacterial coinfections of the respiratory tract. Biochemistry (Moscow). 81 (11), 1340‒1349.
Perico L., Benigni A., Casiraghi F., Ng L.F.P., Renia L., Remuzzi G. 2021. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 17 (1), 46‒64.
Latreille E., Lee W.L. 2021. Interactions of influenza and sars-cov-2 with the lung endothelium: Similarities, differences, and implications for therapy. Viruses. 13 (2), 161.
Aydin Ozgur B., Coskunpinar E., Bilgic Gazioglu S., Yilmaz A., Musteri Oltulu Y., Cakmakoglu B., Deniz G., Gurol A.O., Yilmaz M.T. 2020. Effects of complement regulators and chemokine receptors in type 2 diabetes. Immunol. Invest. 50 (5), 478‒491.
Funding
This work was supported by the Russian Science Foundation (project no. 22-24-00987).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This work does not contain any studies involving human and animal subjects.
CONFLICT OF INTEREST
The authors of this work declare that they have no conflicts of interest.
Additional information
Translated by T. Tkacheva
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Uvarova, A.N., Tkachenko, E.A., Stasevich, E.M. et al. The rs2564978(T) Allele Associated with Severe Influenza A Disrupts the Binding Site for Myeloid Differentiation Factor PU.1 and Reduces CD55/DAF Gene Promoter Activity in Macrophages. Mol Biol 58, 255–265 (2024). https://doi.org/10.1134/S002689332402016X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S002689332402016X