
Abstract
Statistical imputation of classical human leukocyte antigen (HLA) alleles is becoming an indispensable tool for fine-map**s of disease association signals from case–control genome-wide association studies. However, most currently available HLA imputation tools are based on European reference populations and are not suitable for direct application to non-European populations. Among the HLA imputation tools, The HIBAG R package is a flexible HLA imputation tool that is equipped with a wide range of population-based classifiers; moreover, HIBAG R enables individual researchers to build custom classifiers. Here, two data sets, each comprising data from healthy Japanese individuals of difference sample sizes, were used to build custom classifiers. HLA imputation accuracy in five HLA classes (HLA-A, HLA-B, HLA-DRB1, HLA-DQB1 and HLA-DPB1) increased from the 82.5–98.8% obtained with the original HIBAG references to 95.2–99.5% with our custom classifiers. A call threshold (CT) of 0.4 is recommended for our Japanese classifiers; in contrast, HIBAG references recommend a CT of 0.5. Finally, our classifiers could be used to identify the risk haplotypes for Japanese narcolepsy with cataplexy, HLA-DRB1*15:01 and HLA-DQB1*06:02, with 100% and 99.7% accuracy, respectively; therefore, these classifiers can be used to supplement the current lack of HLA genoty** data in widely available genome-wide association study data sets.
Similar content being viewed by others

Introduction
Human Leukocyte Antigen (HLA) represents the primary system, which encodes cell-surface protein that presents the specific antigen peptides for the host immune system. Specific HLA proteins have been implicated as the major susceptible factors for adverse drug reactions, transplant/graft rejection and a wide range of autoimmune and infectious diseases. The HLA, or major histocompatibility complex (MHC), region in humans is located on chromosome 6p21.3 HLA loci, and HLA proteins are highly polymorphic because of natural selection against a wide range of pathogens.1 For example, as of October 2014, the IMGT/HLA database contained up to 12 000 HLA alleles,2 and HLA-B and HLA-DRB were the most polymorphic in HLA class I and HLA class II genes, with 3693 and 1684 two-field alleles, respectively.
Over the years, different methodologies have been developed for genoty** HLA alleles, from classical two-digit seroty** to four-or-more-digit DNA-based ty** methods. However, HLA genoty** is still notorious for being time consuming and costly for research studies that involve thousands of samples. To overcome these problems, methods for predicting HLA genotypes based on single nucleotide polymorphisms (SNPs) have been developed.3, 4 However, the utility of such prediction methods is limited to specific populations for which a particular prediction system is built. An alternative method uses multiple SNPs in the proximity of HLA regions to predict HLA genotypes. Leslie et al.5 developed an HLA prediction system based on identity-by-descents model; this system uses multiple SNPs to infer haplotype information. Using the same statistical algorithm Dilthey et al.6 developed an integrative software program, HLA*IMP, based on SNP data from European populations with a modification of the SNP selection process, which increased the imputation accuracy. A subsequently developed software program, HLA*IMP:02,7 based on SNP data from multiple populations that can accommodate haplotypic heterogeneity, is also available. Each version of HLA*IMP required users to upload the genotype data to a secure, online server; this requirement may exclude certain research groups from using HLA*IMP.
SNP2HLA is an HLA and amino-acid imputation software program built based on the imputation algorithm used for the software package BEAGLE.8 SNP2HLA has enabled researchers to interrogate functional coding variants within HLA genes that might be causal for certain diseases. Non-synonymous changes within HLA genes might cause variations in the binding affinity of the respective HLA protein, but the exact underlying mechanisms of how such changes contribute to disease susceptibilities remains unknown.
The HIBAG R package is an another tool for HLA genotype imputation based on the attribute bagging method.9 Attribute bagging maximizes the advantages of bootstrap aggregation and the random variables selection methods to improve accuracy of HLA imputation.10 In brief, ensemble classifiers are built by randomly selecting sets of individuals from a training data set and randomly selecting representative SNP markers from a set of available SNP sets. The ensemble classifiers are then used as references for imputation based on an independent research dataset.
HIBAG differs from other HLA imputation software because it only assumes minimal Hardy–Weinberg equilibrium, and HIBAG has proven to be robust for populations with complex linkage disequilibrium blocks that deviate from Hardy–Weinberg equilibrium. In contrast with HLA*IMP and HLA*IMP:02, HIBAG utilizes unphased genotype data directly available from genome-wide association studies SNP panels, shortening the computational phasing steps and eliminating the variation produced by different phasing software packages. HIBAG has, for example, helped to identify novel independent risk HLA alleles for Sjögren's syndrome11 and contributed to the confirmation of which HLA alleles among those that increase the risk of multiple sclerosis was associated with a decreased risk of schizophrenia.12
For HLA genotype imputation with a specific population (for example, the Japanese population), it is essential to build custom population training data sets that include rare HLA genotypes that are confined to the respective population. Here, we determined the overall HLA imputation accuracy attained when using each of two sets of published parameters (HIBAG ASIAN ancestry model or HIBAG multi-ethnic model), as references and validation data sets comprising two groups of healthy Japanese individuals. In addition, functions built into the HIBAG R package were used to generate two custom Japanese population parameter estimates with different sample sizes, and a comprehensive comparison was performed to assess the HLA genotype imputation accuracy across three different genoty** platforms and with different training data sets of different sizes. Further assessment of imputation accuracy was carried out using data from the Japanese narcolepsy with cataplexy patient group in which almost 100% of the Japanese patients carried a specific HLA haplotype; this genetic uniformity makes narcolepsy with cataplexy a good model for HLA imputation assessment.
Materials and methods
Table 1 list the numbers of individuals with four-digit HLA genotypes and numbers of unique HLA alleles for samples from the following three groups: Tokyo Healthy Control (THC), Japan PGx Data Science Consortium (JPDSC), Japanese narcolepsy with cataplexy and HIBAG published references. Each data set is described below.
THC
THC samples were taken from healthy Japanese individuals residing in the Tokyo area. SNP data from the extended MHC region were extracted from three genoty** platforms: Affymetrix 6.0, Affymetrix Axiom, or Illumina HumanOmni BeadChip 2.5. SNPs that failed quality control criteria (minor allele frequency<0.05, call rate<95%, Hardy–Weinberg equilibrium<0.001) were removed; 1905, 5217 and 6245 SNP markers from the extended MHC region were remained for reference building in the Affymetrix 6.0, Affymetrix Axiom or Illumina HumanOmni 2.5 platforms, respectively.
The Luminex Multi-Analyte Profiling System and WAKFlow HLA ty** kits (Wakunaga, Hiroshima, Japan) were used to type five HLA loci (HLA-A, B, DRB1, DQB1 and DPB1), at four-digit resolution.
JPDSC
SNPs and four-digit HLA genotypes were obtained for JPDSC samples, which were collected from healthy Japanese individuals registered with the JPDSC (http://www.jpdsc.org/). Data for SNP markers within the extended MHC regions were extracted from data generated with the Illumina HumanOmni 2.5 platform; after quality control (minor allele frequency<0.05, call rate<99%, Hardy–Weinberg equilibrium<0.0001), 11 145 candidate SNP markers were represented in the final data set.
Luminex Multi-Analyte Profiling System and WAKFlow HLA ty** kits (Wakunaga) were used to type five HLA loci (HLA-A, B, C, DRB1 and DPB1) at four-digit resolution.
Japanese narcolepsy with cataplexy
Japanese narcolepsy with cataplexy samples were obtained by the Neuropsychiatric Research Institute of Japan. As part of the diagnosis criteria, the HLA-B, HLA-DQB1 and HLA-DPB1 loci in each sample were typed at four-digit resolution; in addition, the HLA-DRB1 locus in a subset of samples was typed based on complete linkage between HLA-DRB1*15:01 and HLA-DQB1*06:02. Almost all Japanese narcolepsy with cataplexy patients have been reported to carry this particular risk haplotype.13, 14 In the data set we used, all Japanese narcolepsy with cataplexy samples carried the HLA-DRB1*15:01 -HLA-DQB1*06:02 haplotype.
Data for SNP markers within the extended MHC regions were extracted from data generated with the Affymetrix 6.0 platform; after quality control (minor allele frequency<0.05, call rate<99%, Hardy–Weinberg equilibrium<0.0001), 1327 candidate marker SNPs could be used for HLA imputation.
HIBAG references
To assess the reference sets available in HIBAG R for four-digit HLA imputation accuracy with Japanese samples, the published Asian ancestry or multi-ethnicity parameters estimates were used as references and THC or JPDSC data were used for validation. All published parameter estimates are based on SNP markers with call rates>90%. All HIBAG reference HLA genotypes were determined by sequence-based ty**, sequence-specific oligonucleotides, sequence-specific primer methods or some combination thereof.9 Genoty** platform-specific parameter estimates were used for HLA imputation on the respective genoty** platform; additional information on these platforms and data sets are available at www.biostat.washington.edu/~bsweir/HIBAG/.
Custom Japanese population-specific parameter estimates
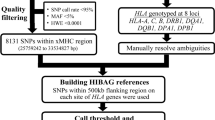
Japanese population-specific parameter estimates were generated based on THC and JPDSC data sets to assess the effect of sample sizes (Table 1) on overall HLA imputation accuracy. HLA imputation accuracy assessments were carried out in following two stages (Figure 1): (1) each data set (THC or JPDSC) was divided randomly into two equal subsets, a training and a validation data set; the result from each training data set served as references for imputation with the respective validation set; and (2) cross-validation using THC/JPDSC data sets in which each data set (THC or JPDSC) as a whole will be used as a training data sets served as reference for imputation with the other validation data set (THC or JPDSC), and the analysis is repeat in a vice versa manner.

Schematic workflow for evaluation of the HIBAG R package.
Imputation parameters
SNPs within the 500 kb flanking regions were used during the SNPs selection process.
HIBAG uses the attribute bagging method for building ensemble classifier. The ensemble classifier parameter was set as 100 bootstrap samples as recommended by the HIBAG author.9
Imputation accuracy assessment in a disease group
SNP variants in disease groups deviate from Hardy–Weinberg equilibrium owing to inflation of risk alleles. Parameter estimates based on THC or JPDSC data were used to assess four-digit HLA imputation accuracy for the Japanese narcolepsy with cataplexy data set in which a specific risk haplotype (HLA-DRB1*15:01 -HLA-DQB1*06:02) is carried by almost every patient represented in the data set.13
Results
The performance of published HIBAG parameters (Asian or multi-ethnic) was evaluated by imputing HLA alleles with two data sets (THC or JPDSC), each comprising healthy Japanese individuals. These same data sets, THC and JPDSC, which differed in sample size, were used to generate custom Japanese-specific parameter estimates. Performances of individual custom Japanese classifiers were evaluated (1) via internal validation in which each data set was divided randomly into training and validation subsets. To assess imputation accuracy, each training data set was used as reference to impute HLA alleles within the respective validation data set and (2) via cross-validation in which each independent data set (THC or JPDSC) was used as the training data set and the other data set was used, in turn, as the validation data set; imputed HLA genotypes were then compared with experimentally determined HLA genotypes. Four-digit HLA imputation performance was mainly represented as prediction accuracy, which was defined as the number of correctly imputed HLA alleles over the total number of imputed HLA alleles. Quality control (call threshold; CT) was applied at CT=0 or CT=0.5 as recommended by HIBAG9 to remove poorly imputed HLA alleles. In addition, a series of CTs was applied to determine the optimum CT for the individual Japanese classifiers while taking accuracy and call rates into consideration. HLA imputation performance for each HLA allele was evaluated based on sensitivity, specificity, positive prediction value and negative prediction value (Supplementary Tables 1–8).
Evaluation of published parameters using THC and JPDSC data sets
Published, genoty** platform-specific parameter estimates, HIBAG Asian ancestry or HIBAG multi-ethnicity, were used as references to impute four-digit HLA genotypes from THC and JPDSC data sets; the imputed genotypes were then compared with experimentally determined HLA genotypes.
THC data generated with three SNP genoty** platforms were used for HLA imputation; the number of markers useful for imputation ranged from 219 to 321 with Affymetrix 6.0, 435 to 805 with Affymetrix Axiom and 573 to 986 with Illumina HumanOmni 2.5 (Table 2). The number of SNP markers used for each classifier depended on the density of SNP markers in the respective genoty** platform; notably, Illumina HumanOmni 2.5 had the highest density of SNP markers in the MHC region among the three platforms. When HIBAG Asian parameters were used as references, four-digit HLA imputation accuracies ranged from 90–96.9% with Affymetrix 6.0, 91.1–98.6% with Affymetrix Axiom and 91.4–98.7% with Illumina HumanOmni 2.5. Among the five HLA loci imputed, HLA-B alleles were consistently imputed with lowest accuracy with data from each genoty** platform; accuracies were 90.5, 91.1 and 91.4% with Affymetrix 6.0, Affymetrix Axiom or Illumina HumanOmni 2.5, respectively (Table 2). When HIBAG multi-ethnic parameters were used, four-digit HLA imputation accuracies decreased slightly to 82.5–96.7% with Affymetrix 6.0, 88.6–98.4% with Affymetrix Axiom and 88.9–98.8% with Illumina HumanOmni 2.5, and imputation accuracies were highest for HLA-DPB1 and HLA-DQB1. In general, HLA imputation with HIBAG references yielded imputation accuracies >90%, and imputation accuracy was generally higher with HIBAG Asian references than with HIBAG multi-ethnicity references.
Using a bigger Japanese control data set (JPDSC) genotyped by Illumina HumanOmni 2.5, the number of SNPs used for imputation at five HLA genes ranged from 260 to 347 for published HIBAG Asian ancestry parameters and 573 to 945 for HIBAG multi-ethnicity published parameters (Table 3). Imputation accuracies ranged from 91.9 to 96.6% using published HIBAG Asian ancestry parameters and from 88.5 to 97.0% using published HIBAG multi-ethnicity parameters. Imputation accuracies were highest for HLA-C and HLA-DPB1 with the HIBAG Asian or HIBAG multi-ethnicity parameters, respectively. With both HIBAG reference sets, imputation accuracy was lowest for HLA-DRB1. The findings were similar to those for the THC validation data set (Table 2); HLA imputation with the JPDSC data set indicated that the HIBAG Asian references outperformed HIBAG multi-ethnicity references for Japanese populations.
Internal validation of imputation accuracy with the THC and JPDSC data sets
Internal validation tests were performed to evaluate the performance of our custom Japanese-based individual classifiers. For each data set, all samples were divided into equal-sized subsets, a training and a validation data set. This random partitioning strategy is one of the functions of HIBAG software in which approximately equal numbers of individuals who carried specific HLA alleles are divided into training and validation data sets.
For THC data without any CT, imputation accuracies observed in the internal validation ranged from 93.0 to 98.3%, 91.7 to 98.3% and 92.2 to 98.3% with Affymetrix 6.0, Affymetrix Axiom or Illumina HumanOmni 2.5 data, respectively. Imputation accuracies increased to 95.3–99.0% (Affymetrix 6.0), 95.1–98.3% (Affymetrix Axiom) and 95.2–99.5% (Illumina HumanOmni 2.5) after applying a CT of 0.5, but call rates were reduced to 84.9–96.6% (Affymetrix 6.0), 88.4–100% (Affymetrix Axiom) or 85.4–97.1% (Illumina HumanOmni 2.5) (Table 4). HLA-B was consistently imputed with the lowest accuracy across the three genoty** platforms, probably because rare HLA-B alleles were underrepresented in the relatively low number of samples in the THC group. Results for HLA imputation accuracy for each specific HLA allele before application of CT are available in Supplementary Tables 1 and after CT application in Supplementary Tables 6–8.
With the JPDSC data set, the internal validation test yielded high imputation accuracy ranges: 95.7–98.9% (CT=0) or 96.7–99.0% (CT=0.5). The call rates fell within the acceptable range of 96.4–99.0% with a CT of 0.5 (Table 4). Results for HLA imputation accuracy at each specific HLA allele before CT application are available in Supplementary Tables 1 and 5, and after CT application in Supplementary Table 9.
Cross-validation test with THC and JPDSC data sets
To evaluate the imputation efficiency of individual classifiers derived from THC or JPDSC data, cross-validation tests were performed in which THC individual classifiers were used as references for HLA imputation with JPDSC data and vice versa (Figure 1). Only overlap** genoty** platform data (Illumina HumanOmni 2.5) and overlap** HLA loci (HLA-A, HLA-B, HLA-DRB1, HLA-DPB1) were used for the cross-validation test. Using THC individual classifiers as reference to impute HLA genotypes from the JPDSC validation data set, we achieved imputation accuracies of 93.5–97.8% across four HLA loci. After applying CT=0.5, accuracies increased to 94.7–98.2% with call rates of 92.6–98.2% (Table 5). Prediction accuracies for HLA-DPB1 were the highest and were closely followed by those for HLA-DRB1, HLA-B and HLA-A. When JPDSC individual classifiers were used as references for imputation with THC data, accuracies were from 95.3 to 98.7%, and accuracies were higher, 96.2–99.1%, after quality control (CT=0.5) with call rates of 95.7–99.5%. Prediction accuracies for HLA-DPB1 and HLA-DRB1 were highest and closely followed by those for HLA-B and HLA-A.
Evaluation of mis-imputed HLA alleles
Four-digit HLA allele imputation accuracies and most likely mis-imputed alleles are shown in Supplementary Table 1. For example, among the HLA-A alleles, HLA-A*01:01, 03:01, 03:02, 31:01 and 33:03 were consistently imputed correctly with all five reference sets. When any one of these alleles was mis-imputed, in most cases, it was imputed correctly to the one-field resolution, for example, when HLA-A*02:01 was mis-imputed, it was always mis-imputed as HLA-A*02:07. This mis-imputed trend was observed in all the six HLA genes we tested (Supplementary Table 1). The consistent pattern of mis-imputation showed the limitation of HLA imputation most likely owing to insufficient samples size at those particular HLA alleles; however, users can refer to Supplementary Table 1 to ascertain the reliability of the imputation accuracy for a particular HLA allele.
CT evaluation
Imputed HLA alleles were filtered by posterior probability (CT) to eliminate poorly imputed HLA alleles. HIBAG instructional materials recommend a CT of 0.5 to remove poorly imputed HLA alleles.9 In using THC data that were genotyped with three different genoty** platforms, we sought to determine the optimal CT for individual Japanese classifiers. Our results (Supplementary Figure 1) showed that a CT of 0.5 was too stringent when compared with a CT of 0.4; accuracy increased only marginally, ~1%, and call rates were reduced by up to ~8%; increasing the CT from 0.4 to 0.5 resulted in a loss of statistical power for identifying associated HLA alleles. We suggested applying a CT of 0.4 when reference data were generated from a relatively homogeneous population, such as a Japanese population.15
HLA imputation test with an independent disease group: Japanese patients presenting narcolepsy with cataplexy
Disease groups are known to have distorted linkage disequilibrium structures relative to normal populations; therefore, we tested the robustness of our custom THC Affymetrix 6.0 individual classifiers, with the Japanese patients presenting narcolepsy with cataplexy disease group. A specific HLA haplotype, HLA-DRB1*15:01 -HLA-DQB1*06:02, is carried by all Japanese patients who present narcolepsy with cataplexy in our data set; therefore, this haplotype is an important supplementary diagnostic indicator for diagnosis of these patients.13, 14 We evaluated the four-digit HLA imputation accuracy with THC references for the available narcolepsy with cataplexy HLA data (HLA-B, HLA-DRB1, HLA-DQB1 and HLA-DPB1). Without any quality control (CT=0), the most accurately imputed HLA locus was HLA-DQB1 (98.6%), followed by HLA-DPB1 (97.2%), HLA-B (94.5%) and HLA-DRB1 (92.4%) (Table 6). With quality control (CT=0.5), imputation accuracies increased for HLA-DQB1 (99.6%), HLA-DPB1 (97.7%), HLA-B (97.0%) and HLA-DRB1 (95.1%); with quality control, call rates decreased and ranged from 69.6 to 97.7% (Table 6). Notably, with our THC references, the HLA-DQB1*06:02 allele was detected with 99.7% accuracy (1 error in 398 samples tested); moreover, HLA-DRB1*15:01 was imputed with 100% accuracy in 86 samples tested, and the remaining 312 samples were imputed as HLA-DRB1*15:01 positive consistent with the HLA profiles of Japanese narcolepsy with cataplexy.13, 14
Discussion
The HIBAG R package,9 together with the published parameter estimates, is freely available to the public. The parameter estimates file only contains information regarding haplotype frequencies with different SNP subsets for imputation. The absence of individual SNP/HLA information in the parameter estimates file encourages the sharing of parameter estimates between researchers without worry of leakage of personally identifiable information. The HIBAG website (www.biostat.washington.edu/~bsweir/HIBAG/) is equipped with an array of parameter estimates, representing the major commercial genoty** platforms stratified by population.
Here, we examine the practicality of using the published parameter estimates with data from a homogeneous population, specifically Japanese populations. We utilized two Japanese healthy control samples sets with different sample sizes to access the imputation accuracy using genotype platform-specific reference in both HIBAG Asian reference and HIBAG multi-ethnic reference. For THC data sets, imputation accuracies ranged from 90.5 to 98.7% with HIBAG Asian references and 82.5 to 98.8% with HIBAG multi-ethnicity references. For the JPDSC data set, imputation accuracies ranged from 91.9 to 96.6% with HIBAG Asian references and 88.5 to 97.0% with HIBAG multi-ethnicity references. In general, HIBAG Asian references outperformed HIBAG multi-ethnicity references with the THC and JPDSC data because of the high representation of rare Asian-specific HLA alleles within the HIBAG Asian reference set in contrast to those of multi population reference set. HLA-DPB1 was a notable exception because the HIBAG multi-ethnic references outperformed the HIBAG Asian references with both the THC and JPDSC data sets at this locus; HLA-DPB1 is relatively less polymorphic than other HLA loci; therefore, the larger number of individuals represented in the multi-ethnicity reference set could increase detection sensitivity for many HLA-DPB1 alleles without sacrificing detection accuracy.
With the built-in function of the HIBAG R package, we used THC and JPDSC data sets to build custom Japanese-based individual classifiers. Using an internal validation method, HLA imputation accuracies of 95.2–99.5% resulted from the THC individual classifiers, and accuracies of 96.7–99.0% resulted from the JPDSC individual classifiers; each accuracy range represented a marked increase from those of the HIBAG Asian or HIBAG multi-ethnicity references. Notably, the JPDSC individual classifiers outperformed the THC individual classifiers mainly because the larger sample size of JPDSC improved the representation of rare HLA alleles. HIBAG9 reported that 10 copies of an allele in the training data set is essential for high imputation sensitivity;9 this requirement may explain the higher accuracy and call rate for the JPDSC data sets. Cross-validation tests showed that near perfect accuracy (99.1%) could be reached for HLA-DRB1 with JPDSC references, imputation accuracies were >96% each for HLA-A, HLA-B and HLA-DPB1 alleles.
Unlike other HLA imputation software, HIBAG uses a variable selection method in which more important/representative SNP markers tend to be used more frequently. And conversely, HIBAG uses SNP markers with lower importance values less frequently, and these markers do not tend to contribute to the accuracy.9 Owing to different quality controls applied by users to genome-wide association study data before HLA imputation, it may be impossible to match a portion of the reliable SNP markers to reference SNPs. Our results showed that, even with a missing-SNP rate as high as 48%, HIBAG predictions were still robust enough to provide imputation accuracies up to 98.1% before CT application and up to 99.1% after applying a CT of 0.5 with only a minor decrease in call rate to 96.7% (Table 5). This result was consistent with previous findings with HIBAG, specifically that missing-SNP rates up to 50% are tolerable.9 In general, a higher density of SNP markers in a MHC region resulted in a higher number of SNP markers being incorporated into an individual classifier, which, in turn, led to more robust predictions in cases of missing SNPs.
Using THC individual classifiers, we successfully imputed risk HLA alleles for narcolepsy with cataplexy with a near perfect accuracy of 99.7% for HLA-DQB1*06:02 and 100% accuracy for HLA-DRB1*15:02. This result indicated that our individual classifiers were robust for detecting risk/protective HLA alleles within a disease group.
However, as with every computational prediction system, accuracy depended on several factors, including (1) the quality of genotype data, (2) the ambiguity of HLA alleles in the reference data set owing to low-resolution HLA ty** and (3) missing SNPs resulting from quality control. Most of the HLA prediction system available does not work well to predict rare HLA alleles but mainly these HLA alleles will be imputed with low posterior probability and would only decrease the call rate of the imputed data and do not increase in false positive of the result.
In summary, although HIBAG is a robust HLA allele prediction system with published parameter estimates that can be used freely by the public, we suggest that custom ensemble classifiers can be built for homogeneous populations, and that sufficient sample sizes are used to build these custom classifiers to achieve higher accuracies of HLA allele imputation. Our individual classifiers built with THC or JPDSC data covered >99% (HLA-A: 99.78%, HLA-C: 99.90%, HLA-B: 99.80%, HLA-DRB1: 99.91% and HLA-DPB1: 99.93%) of the HLA distribution in the Japanese database (www.hla.or.jp), which represents >20 000 Japanese individuals. Our results showed that a CT of 0.4 was sufficient to eliminate poorly imputed HLA alleles. The imputed results should be used as a preliminary screening for existing genome-wide association study data sets, and putative associations should be confirmed via subsequent experiments.
Resources
Japanese individual classifiers would be available on request.
References
Barreiro LB, Quintana-Murci L . From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 2010; 11: 17–30.
Robinson J, Halliwell JA, McWilliam H, Lopez R, Parham P, Marsh SG . The IMGT/HLA database. Nucleic Acids Res 2013; 41: D1222–D1227.
de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 2006; 38: 1166–1172.
Evseeva I, Nicodemus KK, Bonilla C, Tonks S, Bodmer WF . Linkage disequilibrium and age of HLA region SNPs in relation to classic HLA gene alleles within Europe. Eur J Hum Genet 2010; 18: 924–932.
Leslie S, Donnelly P, McVean G . A statistical method for predicting classical HLA alleles from SNP data. Am J Hum Genet 2008; 82: 48–56.
Dilthey AT, Moutsianas L, Leslie S, McVean G . HLA*IMP—an integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics 2011; 27: 968–972.
Dilthey A, Leslie S, Moutsianas L, Shen J, Cox C, Nelson MR et al. Multi-population classical HLA type imputation. PLoS Comput Biol 2013; 9: e1002877.
Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 2013; 8: e64683.
Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR et al. HIBAG-HLA genotype imputation with attribute bagging. Pharmacogenomics J 2013; 14: 192–200.
RG-O Robert Brylla . Francis Quek. Attribute bagging: improving accuracy of classifier ensembles by using random feature subsets. Pattern Recogn 2003; 36: 1291–1302.
Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren's syndrome. Nat Genet 2013; 45: 1284–1292.
Andreassen OA, Harbo HF, Wang Y, Thompson WK, Schork AJ, Mattingsdal M et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol Psychiatry 2014.
Juji T, Satake M, Honda Y, Doi Y . HLA antigens in Japanese patients with narcolepsy. All the patients were DR2 positive. Tissue Antigens 1984; 24: 316–319.
Mignot E, Lin L, Rogers W, Honda Y, Qiu X, Lin X et al. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am J Hum Genet 2001; 68: 686–699.
Paschou P, Ziv E, Burchard EG, Choudhry S, Rodriguez-Cintron W, Mahoney MW et al. PCA-correlated SNPs for structure identification in worldwide human populations. PLoS Genet 2007; 3: 1672–1686.
Acknowledgements
This study was supported by Grants-in-Aid for Scientific Research on Innovative Areas (22133008, KT) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website .
Supplementary information
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Khor, SS., Yang, W., Kawashima, M. et al. High-accuracy imputation for HLA class I and II genes based on high-resolution SNP data of population-specific references. Pharmacogenomics J 15, 530–537 (2015). https://doi.org/10.1038/tpj.2015.4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2015.4
- Springer Nature Limited
This article is cited by
-
In Silico Pipeline to Identify Tumor-Specific Antigens for Cancer Immunotherapy Using Exome Sequencing Data
Phenomics (2023)
-
Protective association of HLA-DPB1*04:01:01 with acute encephalopathy with biphasic seizures and late reduced diffusion identified by HLA imputation
Genes & Immunity (2022)
-
An updated view of the pathogenesis of steroid-sensitive nephrotic syndrome
Pediatric Nephrology (2022)
-
Effect of HLA genotype on intravesical recurrence after bacillus Calmette–Guérin therapy for non-muscle-invasive bladder cancer
Cancer Immunology, Immunotherapy (2022)
-
Importance of HBsAg recognition by HLA molecules as revealed by responsiveness to different hepatitis B vaccines
Scientific Reports (2021)