
Abstract
Caspases work as a double-edged sword in maintaining cell homeostasis. Highly regulated caspase activities are essential during animal development, but dysregulation might lead to different diseases, e.g. extreme caspase activation is known to promote neurodegeneration. At present, visualization of caspase activation has mostly remained at the cellular level, in part due to a lack of cell-permeable imaging probes capable of direct, real-time investigations of endogenous caspase activities in deep tissues. Herein, we report a suite of two-photon, small molecule/peptide probes which enable sensitive and dynamic imaging of individual caspase activities in neurodegenerative models under physiological conditions. With no apparent toxicity and the ability of imaging endogenous caspases both in different subcellular organelles of mammalian cells and in brain tissues, these probes serve as complementary tools to conventional histological analysis. They should facilitate future explorations of caspases at molecular, cellular and organism levels and inspire development of novel two-photon probes against other enzymes.
Similar content being viewed by others

Introduction
Caspases are aspartate-specific cysteine proteases that have attracted significant attention because of their vital roles in apoptosis and inflammation1,2. Dysregulation of these enzymes may cause a variety of diseases including neurodegenerative diseases1,2,3,4. For example, ischemic stroke was the first neurologic disease in which the activation of caspase-1 was documented3,5. In addition to such acute neurologic disease, there is also increasing evidence that suggests caspase-mediated apoptotic pathways might play dominant roles in chronic neurodegenerative diseases including Alzheimer’s and Parkinson’s disease3,4. When, where and how these enzymes are activated are however not clearly understood, especially at the tissue and organism levels. This is in part due to a lack of tools capable of directly imaging them in real time from deep tissues and small animals. Meanwhile, proteolytic activities are usually tightly regulated by physical compartmentalization of the protease in different subcellular organelles, as well as the presence of specific endogenous inhibitor(s)6,7,8. Traditional methods such as immunoassays and colorimetric/fluorimetric assays survey total expression levels and activities of caspases in vitro, but do not provide accurate information about their dynamics9. Consequently, controversies often exist in the precise interpretation of spatial-temporal activation of endogenous caspases as well as the exact subcellular distribution of active caspases10,11,12.
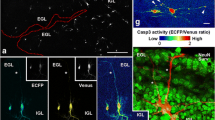
Fluorescence microscopy stands out with its ability to monitor biological targets/processes in native cellular environments with temporal and spatial resolution13. For example, FLICA (Fluorochrome-Labeled Inhibitor of Caspase) is commercially available to assess caspase activities in cells14. Since a FLICA probe itself is fluorescent, “no-wash” imaging is not possible, making it ill-suited for real-time applications. To circumvent this problem, quenched activity-based probes (qABPs) have been developed15,16. The fluorescence of such probes is quenched internally and will be “Turned-ON” only upon the interaction with active caspases. The covalent modification of qABPs on their targets, however, renders such methods limited in their sensitivity and downstream pathway exploration17. Substrate-based imaging probes, based on fluorescent proteins (FPs) or small molecules possessing aggregation-dependent fluorescence properties, provide a direct and real-time measurement of caspase activation with the ability of signal amplification18,19,20,21,22. Genetic manipulation with FPs and the two-step activation required to form fluorescent aggregates in the latter make these approaches ineffective for rapid imaging of endogenous caspase activation. By virtue of their chemical tractability (e.g., different molecular structures/designs can be installed) and cell permeability, small molecule-based probes capable of being rapidly “Turned-On” in a single step in caspase activity-dependent manner, are highly desirable, especially if they can also be readily converted into organelle-specific probes for subcellular investigations of caspase activation15. Another problem in caspase probe development is the limited availability of highly selective/sensitive probes towards individual caspases. Such problem is even more severe for caspase-1, which is well-known to have a low expression level and a short half-life in most cells and tissues, and therefore is often overlooked in the presence of other caspases (e.g., caspase-3/-7)23. As such, recent research efforts have primarily focused on using optimized peptide/peptoid sequences for improved substrate specificity, but only a few of them were successfully applied in fluorescence imaging of endogenous caspase activities16,24,25,26. In a process referred to as “reverse design”, Pralnacasan (a non-peptide small molecule caspase-1 inhibitor; Fig. 1) was converted into a bioluminescent probe which exhibited ~1000-fold increase in sensitivity compared to the commercially available fluorogenic peptide substrate27. A coupled protein-probe engineering approach with mutant caspases was recently proposed to further improve the sensitive and specific detection of caspase-1Photophysical and biochemical properties of probes To establish that these newly developed two-photon enzymatic probes were indeed suitable for in situ imaging, we first investigated their photophysical and biochemical properties in vitro (Fig. 2). Under physiological conditions, the parental reporter AAN showed an emission maximum at 496 nm, whereas the dye-coupled probes (i.e. all probes except C1FS) exhibited a shift to ~448 nm (Fig. 2a and Supplementary Fig. 1c). The blue shift in λem likely came from changes in ICT across the conjugated naphthalene π-system caused by conversion of the electron-rich amine to an amide29. The neighboring peptide WHs having different amino acid side-chains may have conferred further quenching effects and produced discrepancy in fluorescence intensity amongst the probes. The fluorescence enhancement properties of these probes upon peptide removal were evident from the difference in quantum yields between AAN (Φ = 0.17) and the probes (Φ = 0.01–0.08). The blue-shifted property of these probes provided an additional advantage that they might be used in ratiometric imaging experiments (Fig. 2b), where simultaneous measurement of two signals resulting from reacted and unreacted forms of the probes in the same sample could be done, thus minimizing a major limitation commonly associated with intensity-based probes. Interestingly, the FRET-based probe C1FS exhibited similar excitation/emission peaks (λex = 330 nm, λem = 462 nm) as its quencher-cleaved product DAN (in the form of a glycine methyl ester; Fig. 2a and Supplementary Fig. 1d). The significantly brighter one- and two-photon fluorescence emission of DAN (ε330·Φ = 7953, δ·Φ = 142.4 GM), when compared to C1FS (ε330·Φ = 116, δ·Φ = 0.3 GM), clearly demonstrated the Turn-ON effect of this probe upon proteolytic release of the quencher (Fig. 2c). We also noted the more favourable fluorescence properties of DAN over AAN (ε344·Φ = 1683, δ·Φ = 37.0 GM), which might be due to a greater delocalization of the lone-pair electrons in the dimethylamino group of DAN that stabilizes the dye’s excited state, indicating C1FS might offer more sensitive caspase-1 detection than C1RS and C1RA/C1RB. (a) Photophysical and kinetic properties of probes and free dyes. All measurements were taken in 50 mM HEPES, 50 mM NaCl, 0.1% CHAPS, 10 mM EDTA, 5% glycerol, 10 mM DTT, at pH = 7.2. (b) Time-dependent ratiometric fluorescence changes of C1RB (6 μM) incubated with caspase-1 (10 nM) over 2 h at room temperature. Data were obtained by plotting the normalized ratio of fluorescence intensity at 500 nm (F500 nm) over that at 450 nm (F450 nm). (Inset) time-dependent fluorescence emission spectra with λex = 350 nm. (c) Time-dependent “Turn-ON” effect of C1FS (6 μM) incubated with caspase-1 (10 nM) measured and normalized at 460 nm (F460 nm) over 2 h at room temperature, with the corresponding fluorescence emission spectra (inset). λex = 350 nm. (d) Normalized S/N ratio of each of the four caspase-1 probes (6 μM) incubated with 25 μg of BV2 lysates spiked with different concentrations of caspase-1 (37 °C, t = 2 h). S and N represent the fluorescence emission intensity of each reaction solution in the presence (S) and absence (N) of caspase-1. (e) Normalized S/N ratio of each of the 3 caspase-1 probes (C1RB/C1RS/C1FS; 6 μM) incubated with different caspases (t = 2 h, room temperature). (−) no caspase; (caspase-1) 1.5 nM; (caspase-3) 0.8 nM; (caspase-6) 0.2 μM; (caspase-8) 10 nM; (caspase-9) 0.5 μM. (f) Probe selectivity profiles with the corresponding commercial tetrapeptide-AMC as references. For each probe (6 μM), uniform conditions with different caspases were applied. Normalized, relative fluorescence (RFU) values were obtained after 2-h incubation at room temperature. For (d–f), λex = 360 ± 40 nm; λem = 528 ± 20 nm, except for tetrapeptide-AMC and C1FS where λem = 460 ± 40 nm. Kinetic and substrate specificity studies with a panel of recombinant caspases under in vitro conditions were next carried out (Fig. 2a,f, Supplementary Table 1 and Supplementary Figs 1i–l and 2); all probes showed the expected caspase-responsive activities, with virtually identical selectivity and kinetic profiles as those obtained from commercial coumarin-based AMC substrates. Both small molecule probes, C1RS and C1FS, were proteolytically stable and did not display any noticeable degradation in BV2 cell lysates over 24 h (Supplementary Fig. 3). Both were also excellent caspase-1 substrates, with Kcat/KM values (84.3 ± 11.1 and 68.4 ± 0.6 mM−1s−1) falling between the two peptide-based substrates C1RA (Ac-YVAD-AAN; Kcat/KM = 2.7 ± 0.3 mM−1s−1) and C1RB (Ac-WEHD-AAN; Kcat/KM = 516.0 ± 17.0 mM−1 s−1). The WEHD sequence was previously shown to be a much better caspase-1 substrate than YVAD48. Of note, C1RB appeared to possess the lowest detection limit in detection caspase-1 activity, whilst C1FS gave the strongest S/N ratio at higher caspase-1 concentrations (>2 nM, Fig. 2d), likely due to its brighter fluorophore (i.e. DAN). In the substrate specificity studies where other caspases were present (Fig. 2e), both C1RS & C1FS showed improved selectivity over C1RB after a 2-h caspase treatment at room temperature, supporting the notion that small molecule probes might possess improved enzyme selectivity over peptide-based probes27. For subsequent two-photon, live-cell bioimaging, we chose C1RB/C1FS, C3RA/C3RM/C3RE and C8RA for caspase-1, -3/-7 and -8, respectively, as these probes possess the most ideal in vitro reactivity and selectivity profiles. We confirmed they were minimally cytotoxic (Supplementary Fig. 4) and cell-permeable (Supplementary Fig. 5a,c). In an artificial caspase-activation cell model, in which active recombinant caspases were individually delivered into NIH/3T3 cells, all probes were capable of imaging their intended caspase activities under “no-wash” conditions (Supplementary Fig. 5b); for caspase-1 detection, C1FS again showed higher contrast than C1RB, further confirming the improved sensitivity in C1FS as observed in our earlier in vitro studies. We next used the probes to image endogenous caspases in well-established apoptosis and neural models (Fig. 3) under two-photon microscopic settings. It was suggested that microglia, the resident immune cells of the central nervous system, play prominent roles in the pathogenesis of neurodegenerative diseases4,49,50. Several caspases, including caspase-3/-7 and -8, were proposed to be involved in the regulation of microglia activation50. BV2 (an immortalized murine microglial cell line) was thus chosen as our neural model for real-time imaging of caspase activation in response to external stimuli. A previous report indicates lipopolysaccharide (LPS) (a ligand for Toll-like receptor 4, or TLR4) causes microglia activation but not cell death via activation of caspase-8-dependent caspase-3/-7 pathways50. We were therefore curious to know how BV2 might respond to other apoptosis-inducing stimulations. Staurosporine (STS), a potent pan-kinase inhibitor, is known to induce caspase-3 activation and apoptosis in a variety of mammalian cells including HeLa cells15,51. In our experiments, both BV2 and HeLa cells treated with STS showed obvious shrinkage in cell morphology and formation of cleaved casapse-3 fragment (p17) (Fig. 3a–c). C3RA was subsequently added to the cell medium followed by direct live-cell imaging; strong intracellular fluorescence was detected in cells after 4-h STS treatment but not in mock cells, and the fluorescence was inhibited by a pan-caspase inhibitor, Z-VAD(OMe)-FMK (Fig. 3a,b, top). When compared to C3RA, the commercial Ac-DEVD-AMC showed significantly poorer fluorescence properties for endogenous imaginging of caspase-3-like activities from live HeLa cells under two-photon settings (Fig. 3d). To detect whether any impromptu caspase-8 activation might have occurred under the same induction conditions, the entire experiment was repeated with C8RA together with the corresponding Western blotting (WB) analysis (Fig. 3a–c and Supplementary Fig. 5e,h); interestingly, while a death stimulus such as STS caused BV2 cells to undergo caspase-8 activation as shown in both the cellular imaging and WB results, no obvious caspase-8 activation was detected in HeLa cells under similar conditions. We further quantitatively analysed caspase activities in the lysates from the same STS-treated cells with the probes (Supplementary Fig. 5d,g); similar fluorescence increase profiles were obtained. To better illustrate the dynamics in caspase activation, real-time imaging of BV2 cells was next carried out (Fig. 3e). Live BV2 cells were pre-incubated with C3RA for 1 h, stimulated with STS then imaged. Caspase-8-like activity was imaged concurrently by using C8RA; fluorescence signals in both C3RA and C8RA channels started to develop in the cytosol of the treated cells within 30 min after STS was added, and slowly increased over the next 1.5 h (Fig. 3f). Consistent with the occurrence of apoptosis, we observed both cell shrinkage and formation of cleaved caspase-3 fragment (p17) (Fig. 3g). However, the same WB analysis carried out by using the corresponding antibody capable of detecting both the inactive uncleaved (p55) and the active fragmented (p43) caspase-8 revealed that no appreciable formation of the cleaved caspase-8 (p43) could be detected in the initial 30 min; this band started to emerge in the WB analysis ONLY at around 60-min time point, before becoming much more prominent at the 120-min time point. This clear discrepancy between our imaging-based and WB results was likely due to the limited sensitivity of the antibody used in the WB analysis, but also underlines the improved sensitivity of our newly developed probes and its potential application in live-cell imaging experiments. Our results herein are thus consistent with previously reported findings50, but offer a complementary method for more sensitive and dynamic imaging of caspase activities in live neural cells. Taken together, we concluded that C3RA and C8RA were indeed suitable for real-time two-photon imaging of endogenous caspase-3/-7 and -8 activities, respectively, in live mammalian cells. TPFM (λex = 760 nm; λem = 480-550 nm for all AAN-probes) of STS-induced (a) BV2 cells (2 μM, 4 h) and (b) HeLa cells (1 μM, 4 h) using C3RA/C8RA (24 μM, 1 h). Panels 3/6: cells pretreated with Z-VAD(OMe)-FMK (50 μM, 1 h) before STS addition. Panels 1/4: normal cells (DMSO). (c) WB analysis of cleaved caspase-3/-8 from (a,b). (d) TPFM of normal/apoptotic Hela cells imaged by C3RA (24 μM, up) or Ac-DEVD-AMC (40 μM, down; λem = 420–500 nm). Panels 1/3: normal cells. Panels 2/4: STS-induced cells (1 μM, 4 h). Bottom inset: bright field. Top inset: one-photon images (λex = 405 nm). (e) Real-time imaging of caspase-3/-8-like activities in live BV2 cells using 24 μM of C3RA (top)/C8RA (bottom) upon STS (2 μM) stimulation over 2 h. (f) Graphical quantification of RFU extracted from (e). (g) WB analysis of time-dependent caspase-3/-8 cleavage from STS-induced (2 μM) BV2 lysates. (h) WB analysis of time-dependent caspase-3/-7/-8 activation by detection of cleaved caspase-3/-7/-8 from lysates of HeLa cells stimulated with STS (2 μM). (i) RFU from real-time imaging of caspase-3-like activity in live HeLa cells with C3RA/C3RM/C3RE (24 μM, added 1 h before STS) upon STS (2 μM) stimulation. (j) Representative images (t = 60 min) from (i). (k) IF of STS-stimulated (2 μM, 2 h) HeLa cells by anti-caspase-3 (p17) antibody (red) with 3D projections of z-stack images at 90° view and merged with MitoTracker (green, left) or ER tracker (cyan, right). (l) TPFM of caspase-1 activation in primary cortical neurons upon glucose deprivation (GD, 8 h) followed by 1-h incubation with C1RB (24 μM) or C1FS (12 μM; λem = 450–550 nm). Inset: bright field images. NC: neuron cells incubated in normal neurobasal medium. Scale bar = 10 μm in all images. As earlier mentioned, the two organelle-directed caspase-3/-7 probes, C3RM and C3RE, were designed to study whether compartmentalized caspase-3/-7 activation could be imaged as both probes were expected to accumulate selectively in their designated organelle, awaiting fluorescence “Turn-ON” by the corresponding caspase activated therein upon internal/external stimuli. Together with the non-organelle-directed probe, C3RA, we were hopeful that these three probes, when used in combination, would help in painting a more dynamic and spatially resolved view of when and where endogenous caspase-3/-7 might be activated, especially when compared to traditional immunoassays as earlier mentioned. It is worth noting that, direct live-cell measurement of the “OFF/ON” state from a pre-localized, caspase-detecting probe should in practice provide a more easily quantifiable means to measure locally activated caspase activities when compared to similar imaging experiments carried out with non-localized probes and organelle-specific fluorescent trackers. Again, STS-treated HeLa cells were used as a model in which mainly caspase-3 but not caspase-7 was activated (Fig. 3h and Supplementary Fig. 5f). Real-time imaging was carried out (Fig. 3i,j); gradual increases in intracellular fluorescence signals were recorded in both C3RA- and C3RM-treated cells within 60 min, while only background fluorescence was detected in C3RE-treated cells. Both the immunofluorescence (IF) with anti-active caspase-3 antibody (Fig. 3k and Supplementary Fig. 6) and WB analysis (Fig. 3h) showed unequivocal activation of endogenous caspase-3. No caspase-7 activation was detected over the same time course. Further co-localization analysis of the IF results indicated the presence of activated caspase-3 in both cytosol and mitochondria, but not in ER (See Supplementary Videos). We thus concluded that upon STS induction in HeLa cells, endogenous active caspase-3 appeared early in the mitochondria but not in the ER. Our results are thus similar to previously reported imaging data obtained from a mitochondria- targeted FP probe for caspase-318. Compared to standard immunoassays, our organelle-directed imaging approach appeared more sensitive (compare Fig. 3h with Fig. 3i at t = 60 min), and offered additional spatial-temporal resolutions in real time. Different from typical regulators of apoptosis, caspase-1 has long been known for its involvement in inflammation, and is primarily responsible for the activation of the key proinflammatory cytokine IL-1β2,23,52. Ischemic stroke was the first neurologic disease in which the activation of caspase-1 was documented and inflammation has also been often implicated in chronic neurodegenerative diseases2,3,4,5. However, the cellular mechanisms of how caspase-1 is involved in these processes remain poorly defined57,58. Seven-day-old C57BL/6 mice were subcutaneously injected with either ethanol or saline (mock control), and sacrificed 8 h later. The 200-μm-thick sections of the brain were dissected and incubated with C3RA, then imaged by TPFM. As shown in Fig. 4a, the Z-axis scanning allowed sensitive, real-time detection of caspase-3/-7-like activities to as deep as 160 μm of the tissues (Panels 1–4). Pre-treatments of the tissues with caspase-3/7 inhibitor I completely abolished the fluorescence. Further processing of TPFM at a single Z-plane (at 120 μm; Panels 5–8) provided quantitative fluorescence readings that showed consistent caspase-3 activation as those obtained from WB analysis and in vitro enzymatic assays with the corresponding tissue lysates (Fig. 4b,c; the signals from the mock controls, due to basal-level expression of caspase-3 activity, was set as “0” in RFU quantification). No obvious caspase-1 and -8 activations were detected in the same tissue samples probed with C1RB and C8RA, respectively, as well as by WB analysis (Fig. 4c,d). The TPFM images of deep tissues, coupled with the relatively low background fluorescence observed in these images, clearly demonstrated the feasibility of these newly developed probes in future imaging-based, real-time experiments in other tissues and small animals. (a) TPFM of caspase-3 activation in brain slices of ethanol-intoxicated 7-day-old C57BL/6 mouse using C3RA (120 μM, 3 h). Top: 3D images of the mouse brain slice taken along the Z-axis from 0 to 160 μm at 10x magnification. Bottom: images taken at a depth of 120 μm at 40x magnification. Panels 1/5/2/6: control slices (saline-treated); panels 2/6/4/8: treated with caspase-3/7 inhibitor I (200 μM, 3 h). Scale bar = 50 μm. (b) RFU readings extracted from (a) (FTPFM, blue bars) versus those from in vitro enzymatic assays using lysates from the same brain slices (RFU, orange bars). The signal from saline controls was set as “0” and that from ethanol-treated samples was set as “1” in RFU quantification. Due to the base-level active caspase-3 in the saline controls, negative readings were shown with inhibitor-treated samples. (c) WB analysis of caspase-3/-1/-8 activation in lysates of the same mouse brain slices shown in (a). (d) Normalized RFU readings from in vitro enzymatic assays using brain lysates (300 μg) of saline/ethanol treated mice after incubation with C1RB/C3RA/C8RA (6 μM) for 1 h at 37 °C.
Bioimaging of active caspases in live cells


Conclusion
In conclusion, we have successfully developed a suite of cell-permeable probes suitable for sensitive and real-time imaging of target caspase activities in classic neurodegenerative models with two-photon fluorescence microscopy. The two-photon capability of these probes were enabled by the use of suitable two-photon dyes (AAN and DAN), and with observed significant fluorescence enhancement upon probe cleavage in response to different caspases. “No-wash” imaging of caspase activation was carried out with the classic STS-induced apoptosis model in live BV2 and HeLa cells. An organelle-directed approach was further developed that enabled successful detection of mitochondria-localized active caspase-3 activities but not in ER within HeLa cells during 1 h. We believe this novel location-based strategy with C3RM/C3RE could be further explored to study the spatial dynamics of endogenous caspase-3/-7 upon different stimuli. With the feature of low background and high sensitivity endowed by the two-photon caspase-1-detecting probes, we have successfully imaged this enzyme in glucose-deprived mouse primary neurons. Finally, with the ability of directly imaging caspase activities from deep tissues, we observed apparent caspase-3 activation in ethanol-intoxicated mouse brains. Our results indicate these probes are useful tools for studying caspase activation in neurologic disease models. Future work will be directed at the development of more selective caspase probes (e.g., caspase-6/-9), and to extend the same design strategy to other classes of biologically important proteases.
Methods
Chemical synthesis
Detailed information is provided in Supplementary Information.
Solid-Phase Peptide Synthesis (SPPS)
The peptide was synthesized on 2-chlorotrityl chloride resin using Irori™ Microkan reactors following previously published proceduress59. Resulting probes were characterized using LC-MS (IT-TOF) and ESI-HRMS.
C1RA (Ac-YVAD-AAN, C35H41N5O9): m/z[M+H]+calcd, 676.2977; ESI-HRMS found, 676.3005.
C1RB (Ac-WEHD-AAN, C40H42N8O10): m/z[M+H]+calcd, 795.3097; ESI-HRMS found, 795.3114.
C3RA (Ac-DEVD-AAN, C32H39N5O12): m/z[M+Na]+calcd, 708.2487; ESI-HRMS found, 708.2503.
C6RA (Ac-VEID-AAN, C34H45N5O10): m/z[M+Na]+calcd, 706.3059; ESI-HRMS found, 706.3058.
C8RA (Ac-IETD-AAN, C33H43N5O11): m/z[M+Na]+calcd, 708.28519; ESI-HRMS found, 708.2863.
C9RA (Ac-LEHD-AAN, C35H43N7O10): m/z[M+Na]+calcd, 744.2964; ESI-HRMS found, 744.2961.
C3RM (TPP-DEVD-AAN, C52H57N5O12P+): m/z[M]+ calcd, 974.3736; ESI-HRMS found, 974.3733.
C3RE (GLIB-DEVD-AAN, Chemical Formula: C57H68ClN9O19S): m/z[M-3H]3− calcd, 415.4607; found, 415.4610.
Preparation of Pralnacasan-derived small molecule probes was briefly introduced as follows.
C1RS: Compound 7 (10.2 mg, 0.014 mmol) dissolved in CH2Cl2 was added TFA (20% v/v) at 0 °C. The reaction was warmed up and further stirred at room temperature for 4 h. The reaction mixture was concentrated, and the resulting residue was purified by flash chromatography (EtOAc:Hexane = 50:1 with 1% acetic acid) to afford C1RS as pale brown solid (3.9 mg; 52% yield). 1H NMR (300 MHz, DMSO-d6): δ 9.05 (d, J = 9.0 Hz, 2H), 8.61–8.52 (m, 3H), 8.32 (s, 1H), 8.12–8.01 (m, 3H), 7.92–7.62 (m, 5H), 5.23 (d, J = 11.5 Hz, 1H), 4.92 (d, J = 23.1 Hz, 1H), 4.66 (d, J = 19.3 Hz, 1H), 4.37 (d, J = 9.5 Hz, 1H), 4.12 (t, J = 7.2 Hz, 1H), 3.92 (d, J = 5.6 Hz, 2H), 2.67 (s, 3H), 2.27 (t, J = 7.2 Hz, 2H), 2.19–2.05 (m, 4H), 1.98 (s, 2H).13C NMR (75 MHz, DMSO-d6): δ 198.0, 175.2, 172.0, 171.6, 170.1, 170.0, 165.9, 150.1, 141.2, 139.3, 137.0, 136.1, 133.3, 131.1, 130.8, 130.4, 129.1, 128.9, 128.0, 127.5, 127.0, 126.0, 124.4, 124.2, 121.1, 115.2, 52.6, 51.1, 48.8, 44.1, 36.2, 30.2, 29.7, 27.0, 26.1, 19.4. ESI-HRMS: m/z[M+Na]+calcd., 701.2330; found, 701.2358.
C1FS: To the solution of F4 (97 mg, 0.12 mmol) in ethanol (10 mL) was added hydrazine hydrate (60 mg, 1.2 mmol). The resulting mixture was stirred at 60 °C for 2 h. Upon solvent evaporation, the residue containing the desired free amine was re-dissolved in DMF (2 mL). To a separate reaction vessel was added DMF (3 mL), 6-(dimethylamino)-2-naphthoic acid (40 mg, 0.18 mmol) and PyBOP (125 mg, 0.24 mmol). The resulting mixture was stirred at room temperature for 10 min. Then the above amine solution was added. The resulting mixture was stirred at room temperature overnight. Upon solvent evaporation, the resulting residue was added water (20 mL) and EtOAc (50 mL). The organic layer was washed with water (3 × 20 mL), brine (20 mL), dried over Na2SO4, filtered and concentrated, before being purified by flash chromatography (EtOAc:Hexane = 10:1 to 1:1) to give the coupling product which was further dissolved in a mixture of CH2Cl2 (5 mL) and TFA (5 mL). The mixture was stirred at room temperature for 8 h. Upon solvent evaporation, the resulting residue was purified by preparative TLC (CH2Cl2:MeOH = 20:1) to give the final product C1FS (11.4 mg, 11.2% yield). 1H NMR (300 MHz, DMSO-d6): δ 8.50 (d, J = 7.5 Hz, 1H), 8.44 (d, J = 7.5 Hz, 1H), 8.36 (m, 2H), 8.31 (brs, 1H), 8.15 (m, 1H), 7.93 (m, 2H), 7.79–7.85 (m, 4H), 7.69 (d, J = 8.5 Hz, 1H), 7.28 (dd, J = 2.5 Hz, J = 9.0 Hz, 1H), 6.95 (d, J = 2.0 Hz, 1H), 6.93 (d, J = 9.5 Hz, 2H), 5.13 (t, J = 5.0 Hz, 1H), 4.87–4.92 (m, 1H), 4.53 (q, J = 7.0 Hz, 1H), 4.41 (m, 1H), 3.12–3.37 (m, 3H) 3.40–3.50 (m, 4H), 3.04 (m, 6H), 2.94 (m, 1H), 2.62 (dd, J = 6.0 Hz, J = 16.0 Hz, 1H), 2.48 (m, 1H), 2.34 (m, 1H), 2.17–2.24 (m, 2H), 2.04 (m, 1H), 1.90 (m, 1H), 1.68 (m, 1H), 1.60 (m, 1H), 1.14 (t, J = 7.0 Hz, 3H).13C NMR (125 MHz, DMSO-d6):δ 172.3, 169.8, 166.9, 156.7, 152.0, 150.0, 147.2, 143.2, 136.7, 130.2, 128.1, 126.8, 126.6, 126.0, 125.4, 125.1, 124.9, 122.9, 117.1, 111.9, 105.4, 52.8, 50.3, 49.2, 49.1, 45.4, 41.4, 37.1, 30.4, 29.2, 25.9, 19.5, 12.5. ESI-HRMS: m/z [M+H]+calcd., 849.3678; found, 849.3716.
In vitro Enzymatic Assay
All enzymatic assays were carried out in HEPES buffer (50 mM Hepes, 50 mM NaCl, 0.1% CHAPS, 10 mM EDTA, 5% glycerol, 10 mM DTT, pH = 7.2) supplemented with 0.02% Triton X-100 at 25 °C for recombinant caspases, and at 37 °C for lysates. Appropriate enzyme/lysate dilutions were added as indicated, to reaction solutions containing our probes at a final concentration of 6 μM in 384-well microplate (Greiner Bio-One #781900). Control experiments with commercial tetrapeptide-AMC substrates were done where indicated. For inhibitor treatments, Z-VAD(OMe)-FMK (50 μM, ab120487, Abcam) or caspase-3/7 inhibitor I (200 μM for tissue lysate, Calbiochem #218826) was pre-incubated with biological samples for 3 h at 4 °C, prior to incubation with the probe. Liberation of fluorescence (λex = 360 ± 40 nm; λem = 528 ± 20 nm for AAN, and λem = 460 ± 40 nm for AMC and DAN) was recorded by using a BioTek Synergy 4 plate reader. Kinetic constants were computed by fitting the data to Michaelis-Menton equation using a non-linear regression via software Origin™. The error bar in graphs or “±” in the table is a standard deviation (s.d.) of the data.
General procedures for one- and two-photon fluorescence imaging of live cells
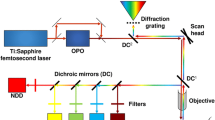
NIH/3T3, HeLa and BV2 cell lines used in our experiments were cultured in DMEM medium supplemented with 10% (for NIH/3T3 and HeLa) or 5% (for BV2) fetal bovine serum (FBS), with 100.0 mg/L streptomycin and 100 IU/mL penicillin. Cells were seeded in glass-bottom dishes (Greiner Bio-One, #627870) and maintained in a humidified atmosphere of 5% CO2 at 37 °C overnight before being imaged. For STS induction, HeLa/BV2 cells were pretreated with/without inhibitor (50 μM Z-VAD(OMe)-FMK) for 1 h before stimulation with STS (LC Laboratories) or an equivalent volume of DMSO for 4 h. C3RA or C8RA (24 μM) was subsequently added and incubation was continued for another 1 h before image acquisition. For real-time imaging, HeLa/BV2 cells in glass-bottom dishes were pre-incubated with the probe for 1 h (C3RM/CERE was washed after 1-h incubation to remove any free probe outside of the targeted organelle), followed by addition of STS (2 μM) and immediate image acquisition (t = 0) over 2–2.5 h. A representative video clip showing real-time imaging was provided in the Supplementary Videos. For one- and two-photon microscopy, data were acquired on a Leica TCS SP5X Confocal Microscope System or on a Carl Zeiss LSM 510 Meta Confocal Microscope as indicated. All 3D images for immunofluorescence were acquired from PerkinElmer Ultraview Vox Spinning Disc confocal microscope and processed with Volocity 6.3.1 3D image analysis software.
TPFM imaging of caspase activities in primary cortical neurons and fresh tissues
All two-photon images were taken on a Leica TCS SP5X Confocal Microscope System. Procedures involving animals were approved by and conformed to the guidelines of Institutional Animal Care and Use Committee at the National Neuroscience Institute (Singapore). Dissociated neuron-enriched cell cultures of cerebral cortex were established from day 16 C57BL/6 mouse embryos, as described53. Primary neurons were cultured in the neurobasal medium (Gibco) with 2% B27 and 0.5 mM GlutaMAX. Experiments were performed in 7–9 day-old cultures. For glucose-deprivation studies, glucose-free neurobasal medium was used (with other components fixed). The cultured neurons were incubated in glucose-free medium for 8 h, while control cells were incubated in normal neurobasal medium. After GD treatment for 8 h, C1RB (24 μM)/C1FS (12 μM) was introduced with further incubation for 1 h. To check whether the signal observed was related to caspase-1 activity, the caspase-1 specific inhibitor, Pralnacasan (50 μM) was added to the cultures 1 h prior to the probe (t = 7 h during GD). Tissues used in the imaging experiments were fresh brains of 7-day-old C57BL/6 mice subcutaneously injected with either ethanol (20% solution in normal saline with 2.5 g/kg at 0 h and again at 2 h) or the same amount of normal saline57,58. At 8 h following the first ethanol dose, the brains were surgically removed from the mouse head and immediately transferred into an ice-artificial cerebrospinal fluid (ACSF; 138.6 mM NaCl, 3.5 mM KCl, 21 mM NaHCO3, 0.6 mM NaH2PO4, 10 mM D-glucose, 1 mM CaCl2 and 3 mM MgCl2). The brain was cut into 200 μm-thick sections using a vibrating blade microtome in ACSF. Slices were incubated with C3RA (120 μM) in ACSF at 37 °C for 3 h before image acquisition. For inhibition experiments, the slices were treated with caspase-3/7 inhibitor I (200 μM) 3 h prior to addition of C3RA. Treated brains were then transferred to poly-L-lysine-coated cover slips and images were acquired at different depths by changing the Z-axis thickness on the microscope.
Additional Information
How to cite this article: Qian, L. et al. Two-Photon Enzymatic Probes Visualizing Sub-cellular/Deep-brain Caspase Activities in Neurodegenerative Models. Sci. Rep. 6, 26385; doi: 10.1038/srep26385 (2016).
References
Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742–758 (2011).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Friedlander, R. M. Mechanisms of disease: Apoptosis and caspases in neurodegenerative diseases. New Engl. J. Med. 348, 1365–1375 (2003).
Heneka, M. T., Kummer, M. P. & Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477 (2014).
Friedlander, R. M. et al. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J. Exp. Med. 185, 933–940 (1997).
Brix, K. & Stöcker, W. Proteases: Structure and Function, New York: Springer-Verlag Wien (2013).
Launay, S. et al. Vital functions for lethal caspases. Oncogene 24, 5137–5148 (2005).
Hunter, A. M., LaCasse, E. C. & Korneluk, R. G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 12, 1543–1568 (2007).
Gurtu, V., Kain, S. R. & Zhang, G. Fluorometric and colorimetric detection of caspase activity associated with apoptosis. Anal. Biochem. 251, 98–102 (1997).
Zhivotovsky, B., Samali, A., Gahm, A. & Orrenius, S. Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ. 6, 644–651 (1999).
van Loo, G. et al. Caspases are not localized in mitochondria during life or death. Cell Death Differ. 9, 1207–1211 (2002).
Chandra, D. & Tang, D. G. Mitochondrially localized active caspase-9 and caspase-3 result mostly from translocation from the cytosol and partly from caspase-mediated activation in the organelle-lack of evidence for Apaf-1-mediated procaspase-9 activation in the mitochondria. J. Biol. Chem. 278, 17408–17420 (2003).
Ueno, T. & Nagano, T. Fluorescent probes for sensing and imaging. Nat. Methods 8, 642–645 (2011).
Bedner, E., Smolewski, P., Amstad, P. & Darzynkiewicz, Z. Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): correlation with DNA fragmentation. Exp. Cell Res. 259, 308–313 (2000).
Hu, M. et al. Multicolor, one- and two-photon imaging of enzymatic activities in live cells with fluorescently quenched activity-based probes (qABPs). J. Am. Chem. Soc. 133, 12009–12020 (2011).
Shaulov-Rotem, Y. et al. A novel quenched fluorescent activity-based probe reveals caspase-3 activity in the endoplasmic reticulum during apoptosis. Chem. Sci. 7, 1322–1337 (2016).
Sanman, L. E. & Bogyo, M. Activity-based profiling of proteases. Annu. Rev. Biochem. 83, 249–273 (2014).
Zhang, Y. et al. Detection of mitochondrial caspase activity in real time in situ in live cells. Microsc. Microanal. 10, 442–448 (2004).
Garrod, K. R. et al. Dissecting T cell contraction in vivo using a genetically encoded reporter of apoptosis. Cell Rep. 2, 1438–1447 (2012).
Shi, H. et al. Real-time monitoring of cell apoptosis and drug screening using fluorescent light-up probe with aggregation-induced emission characteristics. J. Am. Chem. Soc. 134, 17972–17981 (2012).
Ye, D. et al. Bioorthogonal cyclization-mediated in situ self-assembly of small-molecule probes for imaging caspase activity in vivo . Nat. Chem. 6, 519–526 (2014).
Poreba, M. et al. Small molecule active site directed tools for studying human caspases. Chem. Rev. 115, 12546–12629 (2015).
Walsh, J. G., Logue, S. E., Lüthi, A. U. & Martin, S. J. Caspase-1 promiscuity is counterbalanced by rapid inactivation of processed enzyme. J. Biol. Chem. 286, 32513–32524 (2011).
Demon, D. et al. Proteome-wide substrate analysis indicates substrate exclusion as a mechanism to generate caspase-7 versus caspase-3 specificity. Mol. Cell. Proteomics 8, 2700–2714 (2009).
Poreba, M. et al. Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ. 21, 1482–1492 (2014).
Vickers, C. J., González-Páez, G. E. & Wolan, D. W. Discovery of a highly selective caspase-3 substrate for imaging live cells. ACS Chem. Biol. 9, 2199–2203 (2014).
Kindermann, M. et al. Selective and sensitive monitoring of caspase-1 activity by a novel bioluminescent activity-based probe. Chem. Biol. 17, 999–1007 (2010).
**ao, J. et al. A coupled protein and probe engineering approach for selective inhibition and activity-based probe labeling of the caspases. J. Am. Chem. Soc. 135, 9130–9138 (2013).
Kim, H. M. & Cho, B. R. Small-molecule two-photon probes for bioimaging applications. Chem. Rev. 115, 5014–5055 (2015).
Li, L., Ge, J., Wu, H., Xu, Q.-H. & Yao, S. Q. Organelle-specific detection of phosphatase activities with two-photon fluorogenic probes in cells and tissues. J. Am. Chem. Soc. 134, 12157–12167 (2012).
Li, L., Shen, X., Xu, Q.-H. & Yao, S. Q. A switchable two-photon membrane tracer capable of imaging membrane-associated protein tyrosine phosphatase activities. Angew. Chem. Int. Edit. 52, 424–428 (2013).
Li, L. et al. A sensitive two-photon probe to selectively detect monoamine oxidase B activity in Parkinson’s disease models. Nat. Commun. 5, 3276 (2014).
Li, L. et al. A small-molecule probe for selective profiling and imaging of monoamine oxidase B activities in models of Parkinson’s disease. 54, 10821–10825 (2015).
Na, Z., Li, L., Uttamchandani, M. & Yao, S. Q. Microarray-guided discovery of two-photon (2P) small molecule probes for live-cell imaging of cysteinyl cathepsin activities. Chem. Commun. 48, 7304–7306 (2012).
Kim, D. et al. Reaction-based two-photon probes for in vitro analysis and cellular imaging of monoamine oxidase activity. Chem. Commun. 48, 6833–6835 (2012).
Mu, J. et al. A small-molecule FRET reporter for the real-time visualization of cell-surface proteolytic enzyme functions. Angew. Chem. Int. Edit. 53, 14357–14362 (2014).
Yan, H. et al. Poly beta-cyclodextrin/TPdye nanomicelle-based two-photon nanoprobe for caspase-3 activation imaging in live cells and tissues. Anal. Chem. 86, 11440–11450 (2014).
Beaudouin, J., Liesche, C., Aschenbrenner, S., Hörner, M. & Eils, R. Caspase-8 cleaves its substrates from the plasma membrane upon CD95-induced apoptosis. Cell Death Differ. 20, 599–610 (2013).
Wang, C. & Youle, R. J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118 (2009).
Galluzzi, L., Pedro, J. M. B.-S. & Kroemer, G. Organelle-specific initiation of cell death. Nat. Cell Biol. 16, 728–736 (2014).
Smith, R. A. J., Porteous, C. M., Gane, A. M. & Murphy, M. P. Delivery of bioactive molecules to mitochondria in vivo . Proc. Natl. Acad. Sci. USA 100, 5407–5412 (2003).
Abu-Gosh, S. E., Kolvazon, N., Tirosh, B., Ringel, I. & Yavin, E. Multiple triphenylphosphonium cations shuttle a hydrophilic peptide into mitochondria. Mol. Pharmaceutics 6, 1138–1144 (2009).
Dickinson, B. C. & Chang, C. J. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J. Am. Chem. Soc. 130, 9638–9639 (2008).
Madak, J. T. & Neamati, N. Membrane permeable lipophilic cations as mitochondrial directing groups. Curr. Top. Med. Chem. 15, 745–766 (2015).
Hambrock, A., Löffler-Walz, C. & Quast, U. Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Brit. J. Pharmacol. 136, 995–1004 (2002).
Shim, S.-H. et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc. Natl. Acad. Sci. USA 109, 13978–13983 (2012).
Arai, S., Lee, S.-C., Zhai, D., Suzuki, M. & Chang, Y. T. A molecular fluorescent probe for targeted visualization of temperature at the endoplasmic reticulum. Sci. Rep. 4, 6701 (2014).
Benkova, B., Lozanov, V., Ivanov, I. P. & Mitev, V. Evaluation of recombinant caspase specificity by competitive substrates. Anal. Biochem. 394, 68–74 (2009).
Hanisch, U.-K. & Kettenmann, H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394 (2007).
Burguillos, M. A. et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324 (2011).
Belmokhtar, C. A., Hillion, J. & Ségal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20, 3354–3362 (2001).
Denes, A., Lopez-Castejon, G. & Brough, D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. 3, e388 (2012).
Fann, D. Y.-W. et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 4, e790 (2013).
Zhang, W.-H. et al. Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc. Natl. Acad. Sci. USA 100, 16012–16017 (2003).
Park, E. K. et al. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm. Res. 56, 45–50 (2007).
D’Amelio, M., Sheng, M. & Cecconi, F. Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci. 35, 700–709 (2012).
Olney, J. W. et al. Ethanol-induced caspase-3 activation in the in vivo develo** mouse brain. Neurobiol. Dis. 9, 205–219 (2002).
Young, C. et al. Ethanol-induced neuronal apoptosis in vivo requires BAX in the develo** mouse brain. Cell Death Differ. 10, 1148–1155 (2003).
Li, J. & Yao, S. Q. “Singapore Green”: a new fluorescent dye for microarray and bioimaging applications. Org. Lett. 11, 405–408 (2009).
Acknowledgements
Financial support was provided by the National Medical Research Council (CBRG/0038/2013), the Ministry of Education (MOE2012-T2-1-116 & MOE2013-T2-1-048) and the National Natural Science Foundation of China (No. 61505076). We thank Mdm. Yan Tong (CBIS, NUS) for imaging processing, **g Zhai (NUS) for preparation of mouse brain sections and Daniel Sim (NUS) for his initial work.
Author information
Authors and Affiliations
Contributions
L.Q., L.L. and S.Q.Y. designed the experiments. L.Q. performed the experiments and analysed the data with assist from C.W.Z. and K.L.L. for the biological experiments and Y.M. for the probe synthesis. N.G. and Q.H.X. assisted with two-photon related experiments. L.Q. and S.Q.Y. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qian, L., Zhang, CW., Mao, Y. et al. Two-Photon Enzymatic Probes Visualizing Sub-cellular/Deep-brain Caspase Activities in Neurodegenerative Models. Sci Rep 6, 26385 (2016). https://doi.org/10.1038/srep26385
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26385
- Springer Nature Limited