
Abstract
Yin Yang 1 (YY1) is a multifunctional transcription factor shown to be critical in a variety of biological processes. Although it is regulated by multiple types of post-translational modifications (PTMs), whether YY1 is methylated, which enzyme methylates YY1 and hence the functional significance of YY1 methylation remains completely unknown. Here we reported the first methyltransferase, SET7/9 (KMT7), capable of methylating YY1 at two highly conserved lysine (K) residues, K173 and K411, located in two distinct domains, one in the central glycine-rich region and the other in the very carboxyl-terminus. Functional studies revealed that SET7/9-mediated YY1 methylation regulated YY1 DNA-binding activity both in vitro and at specific genomic loci in cultured cells. Consistently, SET7/9-mediated YY1 methylation was shown to involve in YY1-regulated gene transcription and cell proliferation. Our findings revealed a novel regulatory strategy, methylation by lysine methyltransferase, imposed on YY1 protein and linked YY1 methylation with its biological functions.
Similar content being viewed by others


Introduction
YY1 is a ubiquitous and multifunctional zinc-finger transcription factor that is involved in a variety of biological processes, including development, cell proliferation and differentiation, DNA repair and apoptosis, among others1,2,3,4,5,6,7,8,9. YY1 is essential for the development of mouse embryo, with ablation of yy1 in mice resulting in embryonic lethality. Specifically, yy1 mutants undergo implantation and induce uterine decidualization but rapidly degenerate around the time of implantation and yy1 heterozygote embryos display severe developmental abnormalities10. Interestingly, mouse embryonic fibroblast (MEF) cells from mice carrying yy1 alleles expressing various amounts of YY1 display a dosage-dependent requirement of YY1 for cell proliferation11. Accordingly, inhibition of YY1 in cultured cells leads to cytokinesis defects and cell cycle arrest11. YY1 was also shown to function in homologous recombination-based DNA repair (HRR), presumably through its interaction with INO80 chromatin-remodeling complex12. The role of YY1 in apoptosis was first suggested based on the observation that YY1 negatively regulates Hdm2-mediated p53 degradation13. Moreover, YY1 itself is cleaved by caspases both in vitro and in vivo in response to apoptotic stimuli. The cleaved YY1 product, but not wild-type protein can modify the apoptotic response to anti-Fas, suggesting that cleaved YY1 plays a positive feedback role during later stages of apoptosis14. Ample studies indicate expression of YY1 is deregulated in different cancers, including prostate cancer, breast cancer, ovarian cancer, brain cancer, osteosarcoma, colon cancer, cervical cancer, large B-cell and follicular lymphoma, acute myeloid leukemia and hepatoblastoma1,2,4,5.
YY1 exerts its biological functions primarily as a sequence-specific DNA binding transcription factor that can activate or repress gene expression. The structural and functional domains of YY1 protein have been well characterized15,16,17. It contains a transactivation domain at its amino-terminus, a repression domain at its central portion and a DNA binding domain constituted of four zinc fingers of the C2H2 type at its carboxyl-terminus. All four fingers have been shown to be required for proper binding to DNA and involved in transcriptional regulation.
Numerous mechanisms have been shown to regulate the function of YY1, such as its associated co-factors, subcellular localization, post-translational modifications including poly(ADP-ribosyl)ation, ubiquitination, acetylation, O-linked glycosylation, S-nitrosation, sumoylation and phosphorylation. YY1 has been shown to be poly(ADP-ribosyl)ated under genotoxic stress, which negatively regulates its affinity with its DNA binding sites18. In 1998, Walowitz et al. demonstrated that YY1 is a substrate for ubiquitination19. However the exact lysine residues modified by ubiquitination were not determined. Recently, several global proteomic studies have revealed multiple ubiquitination sites including lysine 25820, 174, 203, 204, 339 and 369 (Cell Signaling Technology), with the enzymes responsible for and the function of these modifications remaining to be explored. More recently, Smurf2 was shown to act as an E3 ubiquitin ligase mediating YY1 ubiquitination and degradation, which suppresses B-cell proliferation and lymphomagenesis21,22. Two histone acetyltransferases (HATs), p300 and PCAF (p300-CBP associated factor), have been shown to acetylate YY1 at its central region, which is required for its fully transcriptional repressor activity. PCAF also acetylates YY1 at its C-terminal DNA-binding domain, which might decrease its DNA binding activity23. In response to glucose stimulation, YY1 is O-GlcNAcylated and glycosylated YY1 is released from the Rb protein and free to bind DNA24. Nitric oxide (NO)-induced YY1 S-nitrosylation inhibits its DNA-binding activity, with a functional implication in tumor cell sensitization to Fas-induced apoptosis25. PIASy, a SUMO E3 ligase, has been shown to sumoylate YY1, which increases its stability and represses its transcriptional activity26. Recently, it was shown that the phosphorylation level of YY1 increased dramatically in mitotic cells, which correlates the loss of YY1 DNA-binding activity in mitosis. Furthermore, three phosphorylation sites, serine 247 (S247), threonine 348 (T348) and 378 (T378), were identified, with T348 and T378 phosphorylation proving to be essential for DNA-binding activity of YY1 in vitro27. The same group then identified the first three kinases capable of phosphorylating YY1. Specifically, Polo-like kinase 1 (Plk1) was shown to phosphorylate YY1 at threonine 39 (T39) both in vitro and in vivo. T39 phosphorylation is regulated during cell cycle and peaks at G2/M28. Casein kinase 2α (CK2α) was the second kinase shown to phosphorylate YY1 at serine 118 (S118) in the transactivation domain of YY1. Functional study revealed that blocking S118 phosphorylation increases the cleavage of YY1 during apoptosis29. More recently, it was reported that Aurora B kinase phosphorylates YY1 in the central glycine/alanine (G/A)-rich region at serine 184 (S184) both in vitro and in vivo, which peaks at G2/M and is rapidly dephosphorylated as the cells enters G1. Importantly, phosphorylation of YY1 at S184 is important for its DNA binding activity30. Despite it has been shown to associate with multiple histone methyltransferases, such as protein arginine N-methyltransferase 1 (PRMT1)31, enhancer of zest homologue 1 (EZH1)32 and enhancer of zest homologue 2 (EZH2)33, methylation of YY1, the enzymes responsible for such methylation and hence its function have not been reported so far.
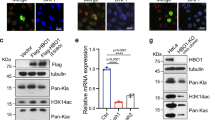
SET7/9 was initially identified as a histone lysine methyltransferase which modifies histone H3 lysine 434,50.
As one of the well-studied functions of YY1 is regulation of cell proliferation, we tested whether SET7/9-mediated YY1 methylation involved in such process. Interestingly, substitution of K173 or K411 to arginine completely abolished the effects of YY1 on promoting cell proliferation (Fig. 6C). Ample studies have shown an intimate link between YY1 function in cell proliferation and tumorigenesis. Therefore, it will be valuable to investigate whether YY1 methylation is enriched and therefore serves as a biomarker in cancers.
Materials and Methods
Plasmids and Cloning Procedures
YY1, SET7/9, G9a, MLL1, SET1A, SET1B, SUV4-20H1, SUV4-20H2, ESET, SUV39H1, SUV39H2, DOT1L, SET8, RIZ1, EZH2 full-length (FL) or truncations were PCR-amplified from cDNA samples prepared from HEK293T cells by using KOD Hot Start DNA Polymerase (Novagen) and then cloned into p3XFLAG-CMV-10 (Sigma), pcDNA3-HA (Invitrogen), pEGFP-C3 (Clontech), pET-28a (+) (Novagen) or pGEX-6P-1 (Promega) expression vectors. All point mutations were generated by using QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene). Luciferase reporter constructs containing YY1 consensus binding motif (CGCTCCCCGGCCATCTTGGCGGCT GGT) or its mutant form (CGCTCCGCGATTATCTTGGCGGCTGGT) were made in pGL2 vector (Promega).
siRNAs, Antibodies, Peptides and Proteins
siRNA specifically targeting YY1 (GACGACGACTACATTGAACAA), SET7/9 (TAGGGCCAGGGTATTATTATA) or LSD1/AOF2 (CTGGAAATGACTATGATTTAA) was purchased from Qiagen. Anti-YY1 K173me1 and anti-YY1 K411me1 antibodies were generated by GenScript, Inc. Antigen (peptide sequence) used for generating anti-YY1 K173me1 and anti-YY1 K411me1 was CSGGGRVK(me1)KGGGKKS and CKSHILTHAKAK(me1)NNQ, respectively; anti-Flag (F1804) antibody was purchased from Sigma; anti-SET7/9 (07–314) was purchased from Upstate; anti-LSD1/AOF2 (A300-215A) was purchased from Bethyl Laboratory, Inc; anti-YY1(H-10) (SC-7341) and anti-GAPDH (SC-25778) was purchased from Santa Cruz Biotechnology. Flag peptide (F-4799) was purchased from Sigma; YY1K173me1 and YY1K411me1 peptides shared the same sequence as the ones used for antibody production.
Plasmids or siRNA Transfection, RNA Isolation and RT-qPCR
Plasmid and siRNA transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Total RNA was isolated using RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. First-strand cDNA synthesis from total RNA was carried out using iScript™ cDNA Synthesis Kit (Bio-Rad), followed by quantitative PCR (qPCR) using Mx3005 machine (Stratagene). All RT-qPCRs were repeated at least three times and representative results were shown. Sequence information for all primers used to check gene expression was presented in Supplementary Table 2.
Protein Immunoblotting and Immunoprecipitation
Protein immunoblotting and immunoprecipitation were performed following the protocol described previously51,52.
Purification of Bacterially-expressed Proteins
His-tagged YY1 full-length (FL), truncations, point mutations were expressed in BL21 (DE3) bacterial cells (Stratagene) and purified by using Ni-NTA agarose (Qiagen), following the protocol described previously51,52.
In vitro Methylation Assay
In vitro methylation assay was performed by mixing purified bacterially-expressed YY1 full length (FL), truncations, point mutations or core histones with histone lysine methyltransferases, either purified from bacterial cells or HEK293T cells with over-expression, in methylation buffer (50 mM Tris-HCl, pH 8.0, 20 mM KCl, 5 mM DTT, 4 mM EDTA) in the presence of 2 μCi L-[methyl-3H]-methionine at 37 °C for 1 hr. The reaction was stopped by adding SDS sample buffer followed by SDS-PAGE gel and autoradiogram. For in vitro methylation assay using peptides as substrates, enzyme (SET7/9) was removed by adding Ni-NTA agarose before dot blot assay.
Generation of SET7/9 Knock-out Cell Lines Using CRISPR/Cas9 Gene Editing Technology
SET7/9 knockout (KO) HeLa cells were generated using CRISPR/Cas9 system as described previously53. gRNA targeting sequence (5′-TAGCGACGACGAGATGGTGG-3′) was cloned into gRNA cloning vector (Addgene, 41824) and confirmed by sequencing. To screen for SET7/9 KO clones, HeLa cells were transfected with pcDNA3.3-hCas9 and gRNA expression vectors (Addgene 41815), followed by G418 selection (0.5 mg/ml). Single colonies were subjected to immunoblotting (IB) using anti-SET7/9 antibody to select knock-out ones, which were further validated by PCR using genomic DNA as template followed by Sanger sequencing. The sequencing information for primer sets used was as follows: Forward (F) 5′-CTCCTCCTCCTCCAAACTCG-3′; Reverse (R) 5′-ACTCCTTCCGCGCTCCAG-3′.
Electrophoretic Mobility Shift Assay (EMSA)
YY1 DNA-binding activity in vitro was examined through EMSA by using the LightShift Chemiluminescent EMSA Kit (20148) from Pierce following manufacturer’s protocol. Briefly, whole cell lysates were prepared in lysis buffer containing 20 mM HEPES, pH 8.0, 1.5 mM MgCl2, 25% glycerol, 420 mM NaCl, 1 mM DTT, 0.4 mM EDTA, followed by repeated freeze-and-thaw cycles and sonication. Each EMSA reaction was set up at a final volume of 20 μl. Around 15 to 30 μg whole cell lysates were mixed with 10 X binding buffer (100 mM Tris, 500 mM KCl, 10 mM DTT; pH 7.5) (2 μl), 50% glycerol (1 μl), 100 mM MgCl2 (1 μl), 1 mg/ml Poly (dI.dC) (1 μl), 1% NP-40 (1 μl) in the presence or absence of unlabeled YY1 consensus binding motif (2 pmol/ μl) (2 μl)(forward: 5′-CGCTCCCCGGCCATCTTGGCGGCTGGT-3′; reverse: 5′- ACCAGCCGCCAAGATGGCCGGGGAGCG-3′) or its mutant form (2 pmol/ μl) (2 μl) (forward: 5′-CGCTCCGCGATTATCTTGGCGGCTGGT-3′; reverse: 5′- ACCAGCCGCCAAGATAATCGCGGAGCG-3′) on ice for 10 mins, followed by incubation at room temperature for 20 mins. Finally, biotin-labeled YY1 consensus binding motif (10 fmol/ μl) (2 μl) (forward: 5′biotin-CGCTCCCCGGCCATCTTGGCGGCTGGT-3′; reverse: 5′biotin-ACCAGCCGCCAAGATGGCCGGGGAGCG-3′) or its mutant form (2 pmol/ μl) (2 μl) (forward: 5′biotin-CGCTCCGCGATTATCTTGGCGGCTGGT-3′; reverse: 5′ biotin-ACCAGCCGCCAAGATAATCGCGGAGCG-3′) was added and incubated at room temperature for 10 mins before adding 5X loading buffer. The protein-DNA complex was resolved by 6% DNA retardation gel (Life Technology, EC63652BOX), followed by electrophoretic transfer to Nylon membrane (Pierce, 77016). The membrane was then cross-linked and subjected to detection using the chemiluminescent nucleic acid detection module (Part No. 89880) provided in the kit following manufacturer’s protocol.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed following the protocol described previously51,52. Briefly, cells were fixed with 1% formaldehyde (Sigma) for 10 mins at room temperature (RT). Fixation was stopped by adding glycine (0.125 M) and incubating for 5 min at room temperature (RT), followed by washing with PBS buffer twice. Chromatin DNA was sheared to 300~500 bp average in size through sonication. Resultant was immunoprecipitated with control IgG or specific antibodies overnight at 4 °C, followed by incubation with protein G magnetic beads (Invitrogen) for an additional 2 hrs. After washing and elution, the protein-DNA complex was reversed by heating at 65 °C overnight. Immunoprecipitated DNA was purified by using QIAquick spin columns (Qiagen) and analyzed by q-PCR using Mx3005 machine (Stratagene). All ChIP-qPCRs were repeated at least three times and representative results were shown. Sequence information for all primers used for ChIP was presented in Supplementary Table 2.
Chromatin Immunoprecipitation Coupled with High Throughput Sequencing (ChIP-Seq)
ChIP-seq sample preparation and computational analysis of ChIP-seq data were performed as described previously with minor modifications51. Library construction: the libraries were constructed following Illumina ChIP-seq Sample prep kit. Briefly, ChIP DNA was end-blunted and added with an ‘A’ base so the adaptors from Illumina with a ‘T’ can ligate on the ends. Then 200–400 bp fragments are gel-isolated and purified. The library was amplified by 18 cycles of PCR. Primary analysis of ChIP-Seq datasets: the image analysis and base calling were performed by using Illumina’s Genome Analysis pipeline. The sequencing reads were aligned to the human genome UCSC build hg18 by using Bowtie2 alignment program. Only uniquely aligned reads were kept. The aligned reads were used for peak finding with HOMER (http://biowhat.ucsd.edu/homer). Of note, promoter (TSS)-associated peaks were defined as those with peak center falling between 100 bp downstream of and 5,000 bp upstream of TSS region. Similarly, enriched motifs in identified peaks were searched using HOMER. YY1 ChIP-seq data were deposited in the Gene Expression Omnibus database under accession GSE69759.
Identification of Proteins Differentially Associated with YY1(wt), YY1(K173R) and YY1(K411R)
To identify proteins differentially associated with YY1(wt), YY1(K173R) and YY1(K411R), whole cell lysates were prepared in lysis buffer containing 20 mM HEPES pH 8.0, 1.5 mM MgCl2, 150 mM NaCl, 25% glycerol, 1 mM DTT, 0.2 mM EDTA, 1% Triton X-100 from HeLa cells stably expressing p3XFLAG-CMV-10-YY1(wt), YY1(K173R) or YY1(K411R) and then subjected to affinity purification by using anti-Flag M2-agarose (Sigma, A2220), washed extensively and eluted with 3XFlag peptides. Elutes were then separated by SDS-PAGE gel followed by silver staining using the SilverQuest Silver Staining Kit (Life Technology, LC6070) following the manufacturer’s protocol. Differentially present bands were subjected to in gel digestion and tandem mass spectroscopy (LC-MS/MS) analysis following the protocol as previously described51.
Global Run-On Coupled to High-Throughput Sequencing (Gro-seq)
Gro-seq was performed following the protocol as described previously51.
Gro-Seq Analysis
The sequencing reads were aligned to hg18 Refseq database by using Bowtie2. EdgeR (http://www.bioconductor.org/packages/release/bioc/html/edgeR.html) was then used to compute the significance of the differential gene expression (P < 0.001). The common artifacts derived from clonal amplification were circumvented by considering maximal three tags from each unique genomic position. Gene ontology analysis for genes regulated by YY1 was done using David54. All Gro-seq data were deposited in the Gene Expression Omnibus database under accession GSE69759.
Luciferase Reporter Assay
HeLa cells were transfected with Renilla luciferase reporter vector (Promega) and luciferase reporter vectors containing YY1 consensus binding site (pGL2-YY1(wt)-luc) or its mutant form (pGL2-YY1(m)-luc) in the presence or absence of YY1(wt), YY1(K173R) or YY1(K411R) for 48 hrs. Cells were then washed with PBS buffer twice and lysed with 1X passive lysis buffer (Promega) before measurement of luciferase reporter activity by using the Dual-Luciferase Reporter Assay System (Promega).
Flow cytometry
Flow cytometry was performed as previously reported52. HeLa cells were trypsinized, washed with PBS and fixed with ethanol at 4 °C overnight. Cells were then washed with PBS and stained with PI/Triton X-100 staining solution (0.1% (v/v) Triton X-100, 0.2 mg/ml DNase-free RNase A (Sigma), 0.02 mg/ml propidium iodide (Roche)) at 37 °C for 15 min. DNA content was then measured and about 105 events were analysed for each sample. Data were analysed using CellQuest (Becton Dickinson) and ModFit LT (Verity Software House).
Cell Proliferation Assay
Cell proliferation assay was performed as previously reported52. Cell viability was measured by using a CellTiter 96 AQueous one solution cell proliferation assay kit (Promega) following the manufacturer’s protocol. Briefly, HeLa cells stably expressing control vector, YY1(wt), YY1(K173R) or YY1(K411R) were seeded at the same density and their proliferation rate was monitored for consecutive two days. Specifically, 20 μl of CellTiter 96 AQueous one solution reagent was added per 100 μl of culture medium and the culture plates were incubated for 1 h at 37 °C in a humidified, 5% CO2 atmosphere incubator. The reaction was stopped by adding 25 μl of 10% SDS. Data was recorded at wavelength 490 nm using a Thermo Multiskan MK3 Microplate Reader.
Additional Information
How to cite this article: Zhang, W.- et al. Regulation of Transcription Factor Yin Yang 1 by SET7/9-mediated Lysine Methylation. Sci. Rep. 6, 21718; doi: 10.1038/srep21718 (2016).
References
Nicholson, S., Whitehouse, H., Naidoo, K. & Byers, R. J. Yin Yang 1 in human cancer. Critical reviews in oncogenesis 16, 245–260 (2011).
Wang, C. C., Chen, J. J. & Yang, P. C. Multifunctional transcription factor YY1: a therapeutic target in human cancer? Expert opinion on therapeutic targets 10, 253–266, 10.1517/14728222.10.2.253 (2006).
Thomas, M. J. & Seto, E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene 236, 197–208 (1999).
Deng, Z., Cao, P., Wan, M. M. & Sui, G. Yin Yang 1: a multifaceted protein beyond a transcription factor. Transcription 1, 81–84, 10.4161/trns.1.2.12375 (2010).
Zhang, Q., Stovall, D. B., Inoue, K. & Sui, G. The oncogenic role of Yin Yang 1. Critical reviews in oncogenesis 16, 163–197 (2011).
Wang, Q. et al. Down-regulation of Sonic hedgehog signaling pathway activity is involved in 5-fluorouracil-induced apoptosis and motility inhibition in Hep3B cells. Acta biochimica et biophysica Sinica 40, 819–829 (2008).
Calame, K. & Atchison, M. YY1 helps to bring loose ends together. Genes & development 21, 1145–1152, 10.1101/gad.1559007 (2007).
Gordon, S., Akopyan, G., Garban, H. & Bonavida, B. Transcription factor YY1: structure, function and therapeutic implications in cancer biology. Oncogene 25, 1125–1142, 10.1038/sj.onc.1209080 (2006).
Castellano, G. et al. The involvement of the transcription factor Yin Yang 1 in cancer development and progression. Cell cycle 8, 1367–1372 (2009).
Donohoe, M. E. et al. Targeted disruption of mouse Yin Yang 1 transcription factor results in peri-implantation lethality. Molecular and cellular biology 19, 7237–7244 (1999).
Affar el, B. et al. Essential dosage-dependent functions of the transcription factor yin yang 1 in late embryonic development and cell cycle progression. Molecular and cellular biology 26, 3565–3581, 10.1128/MCB.26.9.3565-3581.2006 (2006).
Wu, S. et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nature structural & molecular biology 14, 1165–1172, 10.1038/nsmb1332 (2007).
Sui, G. et al. Yin Yang 1 is a negative regulator of p53. Cell 117, 859–872, 10.1016/j.cell.2004.06.004 (2004).
Krippner-Heidenreich, A. et al. Caspase-dependent regulation and subcellular redistribution of the transcriptional modulator YY1 during apoptosis. Molecular and cellular biology 25, 3704–3714, 10.1128/MCB.25.9.3704-3714.2005 (2005).
Bushmeyer, S., Park, K. & Atchison, M. L. Characterization of functional domains within the multifunctional transcription factor, YY1. The Journal of biological chemistry 270, 30213–30220 (1995).
Austen, M. et al. Regulation of cell growth by the Myc-Max-Mad network: role of Mad proteins and YY1. Current topics in microbiology and immunology 224, 123–130 (1997).
Austen, M., Luscher, B. & Luscher-Firzlaff, J. M. Characterization of the transcriptional regulator YY1. The bipartite transactivation domain is independent of interaction with the TATA box-binding protein, transcription factor IIB, TAFII55, or cAMP-responsive element-binding protein (CPB)-binding protein. The Journal of biological chemistry 272, 1709–1717 (1997).
Oei, S. L., Griesenbeck, J., Schweiger, M. & Ziegler, M. Regulation of RNA polymerase II-dependent transcription by poly(ADP-ribosyl)ation of transcription factors. The Journal of biological chemistry 273, 31644–31647 (1998).
Walowitz, J. L., Bradley, M. E., Chen, S. & Lee, T. Proteolytic regulation of the zinc finger transcription factor YY1, a repressor of muscle-restricted gene expression. The Journal of biological chemistry 273, 6656–6661 (1998).
Kim, W. et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Molecular cell 44, 325–340, 10.1016/j.molcel.2011.08.025 (2011).
Ramkumar, C. et al. Smurf2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nature communications 4, 2598, 10.1038/ncomms3598 (2013).
Jeong, H. M., Lee, S. H., Yum, J., Yeo, C. Y. & Lee, K. Y. Smurf2 regulates the degradation of YY1. Biochimica et biophysica acta 1843, 2005–2011, 10.1016/j.bbamcr.2014.04.023 (2014).
Yao, Y. L., Yang, W. M. & Seto, E. Regulation of transcription factor YY1 by acetylation and deacetylation. Molecular and cellular biology 21, 5979–5991 (2001).
Hiromura, M. et al. YY1 is regulated by O-linked N-acetylglucosaminylation (O-glcNAcylation). The Journal of biological chemistry 278, 14046–14052, 10.1074/jbc.M300789200 (2003).
Hongo, F. et al. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochemical and biophysical research communications 336, 692–701, 10.1016/j.bbrc.2005.08.150 (2005).
Deng, Z., Wan, M. & Sui, G. PIASy-mediated sumoylation of Yin Yang 1 depends on their interaction but not the RING finger. Molecular and cellular biology 27, 3780–3792, 10.1128/MCB.01761-06 (2007).
Rizkallah, R. & Hurt, M. M. Regulation of the transcription factor YY1 in mitosis through phosphorylation of its DNA-binding domain. Molecular biology of the cell 20, 4766–4776, 10.1091/mbc.E09-04-0264 (2009).
Rizkallah, R., Alexander, K. E., Kassardjian, A., Luscher, B. & Hurt, M. M. The transcription factor YY1 is a substrate for Polo-like kinase 1 at the G2/M transition of the cell cycle. PloS one 6, e15928, 10.1371/journal.pone.0015928 (2011).
Riman, S. et al. Phosphorylation of the transcription factor YY1 by CK2alpha prevents cleavage by caspase 7 during apoptosis. Molecular and cellular biology 32, 797–807, 10.1128/MCB.06466-11 (2012).
Kassardjian, A. et al. The transcription factor YY1 is a novel substrate for Aurora B kinase at G2/M transition of the cell cycle. PloS one 7, e50645, 10.1371/journal.pone.0050645 (2012).
Rezai-Zadeh, N. et al. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes & development 17, 1019–1029, 10.1101/gad.1068003 (2003).
O’Carroll, D. et al. The polycomb-group gene Ezh2 is required for early mouse development. Molecular and cellular biology 21, 4330–4336, 10.1128/MCB.21.13.4330-4336.2001 (2001).
Wang, L. et al. Hierarchical recruitment of polycomb group silencing complexes. Molecular cell 14, 637–646, 10.1016/j.molcel.2004.05.009 (2004).
Wang, H. et al. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Molecular cell 8, 1207–1217 (2001).
**ao, B. et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 421, 652–656, 10.1038/nature01378 (2003).
Pradhan, S., Chin, H. G., Esteve, P. O. & Jacobsen, S. E. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics : official journal of the DNA Methylation Society 4, 383–387 (2009).
Keating, S. T. & El-Osta, A. Transcriptional regulation by the Set7 lysine methyltransferase. Epigenetics : official journal of the DNA Methylation Society 8, 361–372, 10.4161/epi.24234 (2013).
Couture, J. F., Collazo, E., Hauk, G. & Trievel, R. C. Structural basis for the methylation site specificity of SET7/9. Nature structural & molecular biology 13, 140–146, 10.1038/nsmb1045 (2006).
Dhayalan, A., Kudithipudi, S., Rathert, P. & Jeltsch, A. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chemistry & biology 18, 111–120, 10.1016/j.chembiol.2010.11.014 (2011).
Wang, L. et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer cell 25, 21–36, 10.1016/j.ccr.2013.12.007 (2014).
Kontaki, H. & Talianidis, I. Lysine methylation regulates E2F1-induced cell death. Molecular cell 39, 152–160, 10.1016/j.molcel.2010.06.006 (2010).
Yang, J. et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proceedings of the National Academy of Sciences of the United States of America 107, 21499–21504, 10.1073/pnas.1016147107 (2010).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953, 10.1016/j.cell.2004.12.012 (2004).
Hyde-DeRuyscher, R. P., Jennings, E. & Shenk, T. DNA binding sites for the transcriptional activator/repressor YY1. Nucleic acids research 23, 4457–4465 (1995).
Yant, S. R. et al. High affinity YY1 binding motifs: identification of two core types (ACAT and CCAT) and distribution of potential binding sites within the human beta globin cluster. Nucleic acids research 23, 4353–4362 (1995).
Core, L. J., Waterfall, J. J. & Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848, 10.1126/science.1162228 (2008).
Nguyen, N., Zhang, X., Olashaw, N. & Seto, E. Molecular cloning and functional characterization of the transcription factor YY2. The Journal of biological chemistry 279, 25927–25934, 10.1074/jbc.M402525200 (2004).
Kurash, J. K. et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Molecular cell 29, 392–400, 10.1016/j.molcel.2007.12.025 (2008).
Li, G. et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98, 10.1016/j.cell.2011.12.014 (2012).
Mifsud, B. et al. Map** long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature genetics, 10.1038/ng.3286 (2015).
Liu, W. et al. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155, 1581–1595, 10.1016/j.cell.2013.10.056 (2013).
Liu, W. et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466, 508–512, 10.1038/nature09272 (2010).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826, 10.1126/science.1232033 (2013).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57, 10.1038/nprot.2008.211 (2009).
Acknowledgements
This work was supported by “Thousand Young Talents Program” Funds, the Fundamental Research Funds for the Central University (2013121036), National Natural Science Foundation of China (31371292, 31422030 and 91440112) and Natural Science Foundation of Fujian Province, China (2015J06007) to W.L. and the Fundamental Research Funds for the Central University to X.W. (20720150056).
Author information
Authors and Affiliations
Contributions
W.L. conceived the project. The experiments were performed by W.L., X.W., T.S., H.X., J.Y., H.S., M.H., X.S., F.W., B.P., R.X. and W.G. Bioinformatics analysis was performed by W.Z. and J.D. W.L. wrote the manuscripts with inputs from X.W., T.S., H.S., M.H., J.Y., B.P., R.X., W.G., W.Z. and H.X.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, Wj., Wu, Xn., Shi, Tt. et al. Regulation of Transcription Factor Yin Yang 1 by SET7/9-mediated Lysine Methylation. Sci Rep 6, 21718 (2016). https://doi.org/10.1038/srep21718
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21718
- Springer Nature Limited
This article is cited by
-
Lysine methylation of transcription factors in cancer
Cell Death & Disease (2019)
-
Biological processes and signal transduction pathways regulated by the protein methyltransferase SETD7 and their significance in cancer
Signal Transduction and Targeted Therapy (2018)
-
Regulation of miR-181a expression in T cell aging
Nature Communications (2018)
-
Methylation of transcription factor YY2 regulates its transcriptional activity and cell proliferation
Cell Discovery (2017)