
Abstract
The application of meta-barcoding, qPCR, and metagenomics to aquatic eukaryotic microbial communities requires knowledge of genomic copy number variability (CNV). CNV may be particularly relevant to functional genes, impacting dosage and expression, yet little is known of the scale and role of CNV in microbial eukaryotes. Here, we quantify CNV of rRNA and a gene involved in Paralytic Shellfish Toxin (PST) synthesis (sxtA4), in 51 strains of 4 Alexandrium (Dinophyceae) species. Genomes varied up to threefold within species and ~7-fold amongst species, with the largest (A. pacificum, 130 ± 1.3 pg cell−1 /~127 Gbp) in the largest size category of any eukaryote. Genomic copy numbers (GCN) of rRNA varied by 6 orders of magnitude amongst Alexandrium (102– 108 copies cell−1) and were significantly related to genome size. Within the population CNV of rRNA was 2 orders of magnitude (105 – 107 cell−1) in 15 isolates from one population, demonstrating that quantitative data based on rRNA genes needs considerable caution in interpretation, even if validated against locally isolated strains. Despite up to 30 years in laboratory culture, rRNA CNV and genome size variability were not correlated with time in culture. Cell volume was only weakly associated with rRNA GCN (20–22% variance explained across dinoflagellates, 4% in Gonyaulacales). GCN of sxtA4 varied from 0–102 copies cell−1, was significantly related to PSTs (ng cell−1), displaying a gene dosage effect modulating PST production. Our data indicate that in dinoflagellates, a major marine eukaryotic group, low-copy functional genes are more reliable and informative targets for quantification of ecological processes than unstable rRNA genes.
Similar content being viewed by others

Introduction
Genomic copy number variation (CNV) is increasingly documented in eukaryotic, bacterial and archaeal genomes [1,2,3,4,5], and represents a major source of intra-specific and population-level genetic variation. The impact of CNV on phenotypic trait expression has been characterised in flowering plants, vertebrates, yeast, and human health research including many model organisms [1,2,3,4,5]. Eukaryote CNVs can lead to increased expression and dosage, providing a potential selective advantage [1, 3, 5,6,7]. Despite its potential importance, the scale and role of CNV in most non-model organisms, including marine microbial eukaryotes, is poorly understood.
While CNV has been reported in marine microbial eukaryotes [8,9,10,11,12,13], and a few studies have indicated rRNA genes could vary in copy numbers or sequences [14, 15], it is still relatively unclear whether CNV has a significant impact on quantitative molecular ecological studies employing meta-barcoding, meta-genomics and qPCR [10]. Quantitative molecular ecology studies of marine protists generally use regions of the rRNA operon for community structure analyses due to the broad coverage of rRNA genes in reference databases, the capacity to resolve taxa, and a high genomic copy number (GCN) in eukaryotes (>102 cell−1) which aids in the detection sensitivity [9,10,11,12]. However, in animals, fungi and plants, rRNA gene copies are variably present, from 102–104 copies cell−1 [9, 10, 16,17,18], with a similar range (103–104 copies cell−1) in diatoms (Stramenopiles) [19]. Other groups of microbial eukaryotes may show greater variation, from 103- 105 copies cell−1 in ciliates (Alveolata) [11], and 102–105 in foraminifera (Rhizaria) [12]. Within most species of microbial eukaryotes, rRNA gene copy numbers are considered to be more stable [19, 20], however, relatively few studies have examined this [11, 12, 19].
Dinoflagellates encompass most harmful algal bloom (HAB) forming taxa, as well as constituting up to 50% of marine microbial eukaryotic biomass, thus are a major constituent of aquatic microbial ecosystems [13]. Genome size varies considerably in dinoflagellates (~1 Gb to >150 Gb) including some of the largest known eukaryotic genomes, larger than the largest animal (lungfish, 130 Gb) and plant (Paris japonica, 149 Gb) genomes [21,22,23,24,25]. Gene duplication and large-scale expansion appear to have occurred amongst dinoflagellate genomes, and coding genes are often present in multiple tandem repeats [26,27,28,29,30]. Genomes of coral symbiont species (Dinophyceae: Symbiodinaceae) show highly dynamic evolution, driven by gene family expansion via both tandem duplication [28, 29] and retroposition [31, 32]. Considerable genome size variation and very large genomes occur in multiple planktonic dinoflagellate orders [33], as well as in other groups of marine microbial eukaryotes such as foraminifera, ciliates, and Amoebozoa [33]. GCN of rRNA genes across much of eukaryotic life are considered broadly correlated with genome size [18]. Such large and dynamic genome sizes suggest substantial CNV may exist in these taxa.
Of marine harmful algal blooms forming taxa, those that produce Paralytic Shellfish Toxins (PSTs) are common and have significant public health and economic implications [34]. PST expression can constitute an inducible defence mechanism in marine dinoflagellates in response to the presence of copepod predators [35]. PSTs are synthesised by the cosmopolitan and common marine dinoflagellates Alexandrium species, Pyrodinium bahamense, Gymnodinium catenatum, and Centrodinum punctatum. Dinoflagellate genes associated with PST biosynthesis (sxt) [36,37,38] possess dinoflagellate features such as a unique 22 bp spliced leader sequence on transcripts, a high GC-content, and eukaryotic poly-A tails [36, 38]. A relatively low proportion of genes (~10–27%) in dinoflagellates are thought to be regulated at the transcriptional level [21, 23, 26], with many genes regulated post-transcriptionally. The role of gene dosage acting on this trait may therefore differ in dinoflagellates from that in more highly transcriptionally regulated taxa. Studies of certain species such as A. minutum and A. ostenfeldii have indicated a correlation may exist between cellular PST content and genomic copies of the PST biosynthetic gene sxtA4 [39,40,41]. Some studies have shown that PST synthesis may not be regulated at the transcriptional level [42, 43]. This gene has been found to vary in GCN across studies, as A. pacificum and A. catenella show ~180–325 copies cell−1 of sxtA4 [40, 44, 45]; while A. minutum and A. ostenfeldii showed fewer copies, at 1.5–11 copies cell−1 [39, 41].The majority of species of Alexandrium had no detectable sxtA4 copies and do not produce PSTs [46]. Thus sxtA4 is a gene with a comparatively lower copy number in dinoflagellates than those that show large scale tandem repeats [26,27,28,29,30]. If consistent across PST-producing species, GCN may constitute a useful marker for in situ ecological analyses of HABs, and potentially for other functional traits governed by genomic dosage.
Because rRNA genes, as compared to coding genes, are likely to be under different selective pressures [47], processes that lead to CNV may differ between them. To determine the impact of CNV on both a functional gene and rRNA barcoding markers, and to examine the role of genome size and time in culture on CNV, we quantified CNV of rRNA genes and sxtA4 in relation to genome size across 51 strains of PST-producing marine dinoflagellate, Alexandrium australiense, A. pacificum, A. catenella and A. minutum. Our selection of strains provided capacity to examine CNV within and between species, in strains maintained in long-term culture, and CNV variance across regions. As diversity analysis employing rRNA genes in particular becomes ubiquitous, we aimed to determine the scale of biases associated with CNV, examine its prevalence across dinoflagellates and indicate potential solutions.
Materials and methods
Culture isolation, maintenance and identification
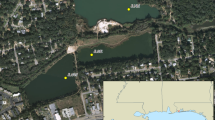
Fifteen non-axenic strains of Alexandrium pacificum were established from a surface net haul collected on 22/11/18 at Mindarie Marina, Western Australia (−31.689127, 115.703103). Single cell isolation of A. pacificum was performed using drawn out glass pipettes and a Nikon Eclipse TS100 inverted microscope (100x magnification). Isolated cells were transferred into Falcon®24 well culture plates containing 1 ml of K/5 medium [48] without sodium silicate. Germanium dioxide was added (5 µg/ml) to prevent diatom growth. Plates were kept at 18 °C under a photon flux of 60–100 μmol photons PAR m−2 s −1 with a 12/12 h dark/light cycle (cool white fluorescent). After 3 weeks, the cultures were transferred into 20 ml K media in 70 mL sterile culture flasks (Thermo Fisher Scientific, Massachusetts, USA), and maintained by serial transfer every 3 weeks. In total 36 additional strains of 4 Alexandrium species (Alexandrium catenella, A. minutum, A. pacificum, A. australiense; Supplementary Table 1) were obtained from collections: the Australian National Algae Culture Collection (CS), the Cawthon Institute Culture Collection of Microalgae (CAWD), the Roscoff Culture Collection (RCC), and collections maintained at the Institute for Marine and Antarctic Studies, University of Tasmania. Strains originated from 8 different countries in Europe, Asia, Australasia and the Americas, across states and regions in Australia, and were isolated on differing dates within the past 30 years.
Isolate identity was confirmed by sequencing the D1-D3 region of large-subunit rRNA. Cells were harvested from 50 ml of culture by centrifugation and DNA extracted using the FastDNA spin kit for soil (MP Biomedicals, Santa Ana, California, USA). DNA quality was checked using a Nanodrop 2000 (Thermo Scientific, Waltham, Massachusetts) spectrophotometer. The D1-D3 region of LSU-rRNA was amplified by PCR using primers D1F [49] and D3B [50] in 25 µl reactions containing 5 µl of 5X MyTaq buffer (Bioline, London, UK), MyTaq polymerase (Bioline, London, UK) 0.5 µl, 7.5 pmol of each primer, 1 µg µl−1 BSA (Biolabs, Arundel, Australia), 1 µl of DNA and 15.5 µl of DNA-free water. PCR conditions were 94 °C for 5 min, followed by 35 cycles of: 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min; and a final extension step of 3 min. PCR products were verified by 1% agarose gel electrophoresis stained with GelRed (Gene Target Solutions, Dural, Australia) and purified with Zymoclean™ (Zymo Research, California, USA). Sanger sequencing of products was performed by Macrogen (South Korea), with strains assigned to A. pacificum, A. catenella, A. minutum or A. australiense based on comparisons with sequences from verified isolates of each species.
Culture synchronisation and harvest
To measure genome size, CNV and PSTs, Alexandrium spp. were grown to exponential phase in GSe medium [51] at 18 °C. For genome size measurement of cell cycle synchronised strains, cells were then incubated for 48 h in darkness to induce synchronisation of cell division [52, 53]. Sub-samples were fixed with Lugol’s iodine at the point of harvest, and cell concentration determined using a Sedgewick-Rafter counting chamber (ProSciTech, Australia) and an inverted light microscope (Leica Microsystems, Wetzlar, Germany). For CNV quantification with qPCR, ~60–75 × 103 cells in triplicate from each strain were harvested by centrifugation (10 min at 1000 g). For genome size quantification using flow cytometry, ~105 cells were harvested in triplicate from each strain. At least 106 cells were harvested by centrifugation for 10 min at 1000 g for PST measurement.
Genome size measurement
Cells were washed with a 1 × PBS, fixed with 1% (w/v) paraformaldehyde for 10 min, and then washed again with 1 × PBS. Cell pellets were re-suspended in 2 mL cold methanol, stored for at least 12 h at 4 °C to remove the intracellular chlorophyll, washed twice with 1x PBS, and then stained for > 3 h in 0.1 mg mL−1 propidium iodide and 2 µg mL−1 RNAse (Merck KGaA, Darmstadt, Germany).
A CytoFLEX S Flow cytometer (Beckman Coulter, California, USA) equipped with laser excitation at 488 nm was used for the flow cytometry analysis. BD™ DNA QC Particles Chicken blood cells (3 pg DNA/nuclei; BD Biosciences, San Jose, USA) were used as a standard [54]. In total, 2-µm fluorescent beads were used as stable particles to verify instrument alignment (BD Biosciences, San Jose, USA). Triplicate samples were run at 30 µL min−1, and data acquired in linear and log modes until at least 10,000 events were measured per sample. Fluorescence emission of propidium iodide stained DNA was detected at 610 ± 10 nm. The peak ratios and coefficient of variation (CV) were quantified with CytExpert software (Beckman Coulter, California, USA). FSC channel was used as a trigger with automatic setting from the manufacturer. Gating and further analysis were only performed for peaks with CV values below 20%, and any peaks above this were rerun. The gating was performed by using FSC-A vs SSC-A gate to exclude debris and use the PI gate on histogram to remove large background noise without the DNA content. Genome size in base pairs used a conversion factor of 1 pg of DNA = 978 Mbp [55].
Genomic copy number quantification with qPCR
DNA extraction was carried out using a PowerSoil DNA Extraction kit (QIAGEN, OH, USA) according to manufacturer’s instructions. DNA was extracted in triplicate, and quality and quantity determined using a Nanodrop ND-1000 (ThermoFisher Scientific, Waltham, Massachusetts) and Qubit 2.0 Fluorometer (ThermoFisher Scientific, Waltham, Massachusetts). qPCR was carried out with a BioRad CFX384 Touch™ System (BioRad, California, USA) using species-specific qPCR assays for Alexandrium rRNA genes [56] and sxtA4 [44] (Supplementary Table 5, Supplementary Fig. 8). qPCRs were run in triplicate using the following cycling parameters: 95 °C for 10 s, 35 replicates of 95 °C for 15 s and 60 °C for 30 s. Total reaction volume was 10 μl, containing 5 μl SybrSelect™ (ThermoFisher Scientific, Massachusetts, USA), 0.5 μM each primer, 1 μl template DNA, and 3 μl PCR-grade water. Samples and Master Mix were loaded to Hard-Shell 384-well PCR plates using epMotion 5075 Liquid Handling Workstations (Eppendorf AG, Hamburg, Germany). Amplification specificity was confirmed using melt-curve analysis. Quantification cycle values were generated by CFX Manager 3.1. Standard curves of sxtA4 and rRNA genes vs quantification cycle (Cq) was developed using ten-fold serial dilution of Gblocks® fragments (Integrated DNA Technologies, Coralville, Iowa, USA). Copy number per µL DNA was determined using the formula:
Standard curves, positive controls and negative controls were included in each sample plate as the sample DNA extracted from Alexandrium strains of known concentration (cells µL−1). Copies of sxtA4 and rRNA genes per µL−1 DNA were determined relative to qPCR standard curves.
Statistical analyses
The significance of relationships between genome size, rRNA gene copies cell−1, sxtA4 copies cell−1 and total PST cell−1 were assessed using Spearman’s rank correlation and linear regression after transformation, as appropriate, as implemented in GraphPad Prism 7.04. Shapiro-Wilk tests were used to examine normal or log normal distributions. Patterns of genome size and rRNA gene CNV associated with cell culture were based on isolation dates provided by culture collections and isolators. Days from isolation date to sample extraction date were calculated. To account for different laboratory growth rates, cultivation days were converted to estimated number of generations based on published growth rates for each species at culture maintenance conditions (50–80 μmoles PAR; 12:12 L:D cycle18–20 °C). Individual strain variance from respective species means were calculated for genome size (pg) and log10 rRNA gene (copies cell−1). Individual strain deviation from species means were calculated using ([(** of picoeucaryotes in marine ecosystems with quantitative PCR of the 18S rRNA gene. FEMS Microbiol Ecol. 2005;52:79–92." href="https://link.springer.com/article/10.1038/s43705-023-00274-0#ref-CR83" id="ref-link-section-d363632177e1365">83, 84].

a Variation in Alexandrium spp genome size with generations in laboratory culture. Values are SD from the mean genome size. Darker background shading indicates range of deviation among Mindarie (WA) Alexandrium pacificum strains cultured for <20 generations prior to analysis. b Variation in Alexandrium species rRNA copy number with increasing generations in laboratory culture. Values are SD from the mean rRNA copy number of each species. Darker background shading indicates range of deviation among Mindarie (WA) Alexandrium pacificum strains cultured for <20 generations prior to analysis.
CNV of rRNA gene and sxtA4 among and within species
rRNA GCN varied by 6 orders of magnitude across Alexandrium species, from 267 ± 18.8 copies cell−1 in a strain of A. minutum (RCC4877) to 1.23 × 108 ± 85 × 106 copies cell−1 in a strain of A. australiense (AT-YC-H) (Fig. 3a). In general, strains of A. australiense and A. pacificum showed the largest rRNA GCN, while those of A.minutum were the smallest and most uniform (Fig. 3a). Within-species CNV of rRNA genes was surprisingly high, particularly among isolates of A. pacificum, which varied from 1.74 × 104 ± 1.26 × 103 copies cell−1 to 1.72 × 107 ± 1.40 × 107 copies cell−1, and more than two orders magnitude (8.1 × 104 ± 1.40 × 104 copies cell−1 to 1.72 × 107 ± 1.41 × 107 copies cell−1) in a single population (Fig. 4). Increased generations in laboratory culture (Fig. 2b, Supplementary Fig. 1B) had no effect on mean rRNA GCN or CNV of Alexandrium species (F = 1.819, df=2, p = 0.177). Variability was evident in sxtA4 copies across Alexandrium species, though at a smaller scale than in rRNA gene (Fig. 3c, Supplementary Fig. 2). The sxtA4 GCN of A. pacificum strains showed the greatest variability, from 7.8 ± 1.9 copies cell−1 to 609 ± 133 copies cell−1, but were more uniform among strains from the same population (43.8 ± 9 copies cell−1 - 609 ± 133 copies cell−1), while the variability of sxtA4 GCN in A. minutum and A. australiense strains were low, from 0–8.9 ± 0.61 copies cell−1 (Fig. 3c). A. catenella strains varied from 19.9 ± 10.5 copies cell−1 - 189.12 ± 58.5 copies cell−1 (Fig. 3c). A significant positive relationship between log10 rRNA copies cell−1 and genome size was evident (Fig. 3b, F = 22.99, df=49, p < 0.0001, r2 = 0.319) across the species of Alexandrium, however at the species level this was only significant within A. pacificum (F = 6.7, df=18, p = 0.0185, r2 = 0.27). A significantly non-zero slope was found in the relationship between log10 sxtA4 copies cell−1 and genome size but explaining only a very low amount of the variance (Supplementary Fig. 2A, F = 5.3, df=43, p = 0.026, r2 = 0.109).

a CNV of rRNA (±SD) in strains of Alexandrium spp. b Relationship between genome size (pg cell−1) and rRNA copies cell−1 in Alexandrium species (F = 22.99, df=49, p < 0.0001, r2 = 0.319). c CNV of sxtA4 (±SD) in Alexandrium species. d Relationship between total PSTs (ng cell−1) and sxtA4 copies cell−1 in Alexandrium species (F = 11.73, df=18, p = 0.003, r2 = 0.395).

a Sites where strains of A. pacificum were isolated from the Pacific region including sites across Australia. b Mindarie from where strains of A. pacificum were isolated. c CNV of sxtA4 (±SD) and rRNA (±SD) in Alexandrium pacificum from all sites and from Mindarie only.
PST content and its relationship to sxtA4 copy number
All strains produced PSTs with the exception of A. australiense ATCJ33 and two strains of A. minutum, RC4874 and CCMI1002. The range of PST congeners produced varied considerably between and within species (Fig. 5). Species of A. catenella contained the highest PST content (Figs. 5, 3d) dominated by C1, 2 and GTX1, GTX2, GTX3 and GTX4. Both GTX5, GTX6, as well as decarboxylated versions of STX and NeoSTX were mostly absent. In contrast, A. minutum strains produced a more restricted range of PSTs, with C2, GTX2 and GTX3 produced (Fig. 5). Total PSTs were more uniform among A. pacificum, with no consistently dominant PST variants. A positive relationship was significant between log10 sxtA4 copies cell−1 and log10 total PSTs ng cell−1 (Fig. 3d, F = 11.73, df=18, p = 0.003, r2 = 0.395). A. minutum produced a higher amount of PST per copy of sxtA4 in comparison to A. pacificum. Strains of A. australiense with extremely low or no detectable PSTs (ATCJ33, AADVN1, AT-YC-H), averaged <1 sxtA4 copy cell−1. In cases where less than one sxtA4 copy cell−1 was found, this was due to copy numbers below the level of quantification of the qPCR assay.

The heatmap shows the inter-species and intra-species variations of each PST congeners' concentration across Alexandrium spp.

