
Abstract
Photoresponsive hydrogels can be employed to coordinate the organization of proteins in three dimensions (3D) and thus to spatiotemporally control their physiochemical properties by light. However, reversible and user-defined tethering of proteins and protein complexes to biomaterials pose a considerable challenge as this is a cumbersome process, which, in many cases, does not support the precise localization of biomolecules in the z direction. Here, we report on the 3D patterning of proteins with polyhistidine tags based on in-situ two-photon lithography. By exploiting a two-photon activatable multivalent chelator head, we established the protein mounting of hydrogels with micrometer precision. In the presence of photosensitizers, a substantially enhanced two-photon activation of the developed tool inside hydrogels was detected, enabling the user-defined 3D protein immobilization in hydrogels with high specificity, micrometer-scale precision, and under mild light doses. Our protein-binding strategy allows the patterning of a wide variety of proteins and offers the possibility to dynamically modify the biofunctional properties of materials at defined subvolumes in 3D.
Similar content being viewed by others

Introduction
Photoresponsive materials benefit from the inherent spatiotemporal control of light to generate sophisticated microstructures1,2. Photoinstructive hydrogels are very promising scaffolds as they offer contact-free remote control of the (bio)physical or (bio)chemical properties by light actuation3,4,5,6,7. Photoresponsive gels have been developed based on photoisomerization8,9,10, photoclick chemistry11,12,13,14,15,16,17, or photolabile protecting groups18,19,20,21,22 in order to modulate their microenvironment by light. Via a contactless mode of action, light allows minute spatial modifications in 2D and 3D as well as temporal control over material features. Applications of photoinstructive hydrogels range from site-specific therapeutic delivery and biosensors to synthetic cell culture platforms for tissue engineering2,23,24. An exciting perspective for protein organization is based on two-photon (2P) patterning techniques25,26. By utilizing femtosecond (fs) pulsed near-infrared (NIR) laser sources, photoreactions can be confined to the focal plane, improving the z resolution to 2–3 µm. Currently, there is considerable interest in using protein-functionalized hydrogels in the context of 3D smart materials, biosensors, and drug delivery systems27,28,29,30. Natural and engineered proteins offer a diverse set of biochemical characteristics and properties. Their integration as biofunctional components into synthetic hydrogel networks is an attractive approach to modulate material features. For instance, hydrogels harboring proteins such as antibodies or enzymes were engineered to change hydrogel properties in response to specific triggers31,32,33. Here, protein arrangement via 2P-patterning of hydrogels facilitates a 3D control over the topography of a photoresponsive material, enabling the creation of hydrogels into more complex 3D devices. In addition, 3D protein immobilization permits a high (bio)specificity over subvolumes of the gel and hence a volumetric control of the response. However, the design and establishment of photoinstructive hydrogels that allow for in-situ protein immobilization are required to dynamically tune the material properties. Due to the lack of selectivity, reversibility, and uniform orientation of protein assembly, 3D protein grafting is far from reaching broad applicability. In particular, the orientation, homogeneity, and stability of proteins immobilized in photoresponsive matrices play a vital role in governing their biochemical properties. Efforts to pattern proteins by 2P-processes are frequently affected by the chemical cross-linking reactions for hydrogel formation, which often involves free radicals, or unspecific bioconjugation resulting in random protein orientation17,22,34. There are only few examples regarding a 3D organization of biomolecules, many of them restricted to synthetic peptides or small molecules15,18,21,22,35,36. Recently, the site-specific patterning of functional proteins by light was achieved via oxime ligation37 and chemoenzymatic strategies38,39,40. Regarding high-precision structuring, advancements in the 2P-patterning specificity and sensitivity at very low laser power are crucial. To overcome these restrictions, generic procedures are required that take advantage of high-affinity protein binders amenable to 2P-patterning of functional proteins in hydrogels.
Here, we developed an optochemical tool based on autoinhibition and photoactivation of multivalent N-nitrilotriacetic acid (trisNTA)41,42. After UV illumination, the photoliberated trisNTA site-specifically binds His6–10-tagged proteins with (sub)nanomolar affinity (KD = 0.1–10 nM, PBS-TE buffer, pH 7.4, 25 °C)43. The ultra-small tag-size combined with the high-affinity binding of His-tagged proteins allow for the direct and oriented immobilization of functional proteins44. In addition, trisNTA offers kinetically stable binding (koff = 0.18 h−1, HBS buffer, pH 7.5, 25 °C) but remains fully reversible in the presence of imidazole, histidine, or EDTA45. Of note, this strategy relies on noncovalent protein association that is less stable compared to covalent protein immobilization37,38,39,40. In live-cell applications, the long-term stability of the trisNTA/His-tag interaction can be affected by the utilized cell media, especially when high histidine concentrations (>200 µM) are present. Nevertheless, the high-affinity trisNTA/His-tag complex formation was successfully exploited in live-cell applications such as in-cell protein labeling, live-cell super-resolution microscopy, or 2D in-situ receptor confinement at single-cell level, demonstrating applicability and sufficient persistence of the interaction pair under cell culture conditions46,47,48.
To conceive a versatile strategy for 3D protein assembly by high-affinity tools, we engineered photoinstructive hydrogels, equipped with a two-photon-activatable trisNTA (TPA-trisNTA) for precise protein organization by 2P laser lithography. By facile addition of 2P-sensitizers, we significantly enhanced the 2P photoscission of TPA-trisNTA regarding efficient protein alignment at low laser power. The improved photoactivation efficacy facilitated the user-defined and site-specific photopatterning of proteins in hydrogels with exquisite control over 3D localization, culminating in high-resolution volumetric structures in multiple z dimensions and 1 µm thin features in x/y dimension. In addition, subcellular protein deposition in close proximity to single cells is demonstrated, providing basic cell applicability.
Results and discussion
Design of a two-photon activatable tool for photopatterning
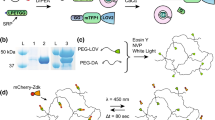
The design of TPA-trisNTA and its functional mechanism is based on the autoinhibition of the multivalent chelator head trisNTA, which is connected via a peptide linker to an intramolecular His5-tag (Fig. 1). In presence of Ni(II) ions, the trisNTA head group is self-inhibited by forming an intramolecular complex (OFF-state). By incorporating a photocleavable amino acid, the inactivating histidines were released upon illumination thereby liberating the trisNTA (ON-state). Due to the high affinity of the trisNTA for His6-10-tagged proteins, the cleaved-off His5-tag is competed out and a stable complex with the protein of interest (POI) is formed. As a result, TPA-trisNTA functionalized matrices are able to immobilize any His-tagged POI with defined orientation after illumination (TRAPPED-state).

a Schematic illustration of the autoinhibition (OFF) and light-activation (ON) of TPA-trisNTA with subsequent trap** of His-tagged POIs. b Design of TPA-trisNTA. The interaction molecule consists of the trisNTA head group and an intramolecular His5-tag at the C-terminus. For 2P photocleavage, a DEAC-based β-amino acid is strategically incorporated within the linker. A single cysteine is used as an anchor for hydrogel immobilization.
For rapid assembly of TPA-trisNTA, we employed fluorenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis (SPPS). To achieve photoactivation of trisNTA by UV and NIR-light, the optochemical tool was strategically equipped with a 7-diethylaminocoumarin (DEAC)-based β-amino acid49. DEAC provides beneficial features e.g., small size, biocompatibility, a strong absorption maximum at 390−400 nm (ε = 20,000 M−1 cm−1), a photolysis quantum yield of up to 0.3 as well as two-photon uncaging properties (Supplementary Fig. 1). A short ethylenediamine carbamate linker was installed, since carbamate protected photocages showed superior light responsiveness and resistance to hydrolysis within hydrogel microenvironments50. To allow hydrogel immobilization, a cysteine residue was included close to the N-terminus. After a six-step synthesis, the DEAC-based β-amino acid was integrated via SPPS into the backbone of TPA-trisNTA (Supplementary Methods 1.1, Supplementary Methods 1.2, Supplementary Fig. 2) and identified after RP-C18-HPLC purification by mass spectrometry (Supplementary Fig. 3, Supplementary Fig. 4).
Photophysical properties of TPA-trisNTA
First, we measured the one-photon absorption and fluorescence spectra of TPA-trisNTA as well as DEAC β-amino acid in HBS buffer/MeCN (1:1) (Fig. 2a, Supplementary Methods 1.4, Supplementary Fig. 5). Both compounds displayed an intense absorption band located around 390 nm and a strong fluorescence emission with a maximum around 480 nm. The absorption profile of TPA-trisNTA compared to the spectrum of the DEAC β-amino acid shows that the light absorbing properties are unaffected.

a Normalized absorption and fluorescence spectra of DEAC β-amino acid and TPA-trisNTA in HBS buffer/MeCN (1:1, v/v). b Quantification of photoscission showed the disappearance of DEAC β-amino acid and formation of the uncaged product as a function of illumination time at 405 nm. c Two-photon absorption cross section values of GM (ϕFϬ2P) for TPA-trisNTA determined by two-photon excited fluorescence measurement and referenced to values of coumarin 307. d Power-dependent fluorescence spectra upon two-photon excitation of TPA-trisNTA at an excitation wavelength of 800 nm. e Intensities of the power dependent fluorescence spectra of TPA-trisNTA were logarithmically plotted against the logarithm of the power. The parameter for two-photon power dependency was found to be 1.93 ± 0.02, confirming a two-photon process for TPA-trisNTA activation at 800 nm.
Next, we verified the one-photon uncaging kinetics of the DEAC-based β-amino acid as a model system upon photolysis. Therefore, the model peptide was illuminated using a 405 nm LED lamp (185 mW/cm2) with increasing light doses (5–900 s) and analyzed by RP-C18-HPLC (Supplementary Fig. 6). The photoscission of the β-amino acid followed a mono-exponential decay with simultaneous mono-exponential formation of the uncaged product, documenting a fast photoactivation (t1/2 = 12 s) under one-photon photolysis conditions (Fig. 2b).
To evaluate the photophysical properties of TPA-trisNTA, the 2P absorption action cross sections (φFϬ2) were determined by 2P-excited fluorescence (TPEF) and given in units of Goeppert-Mayer (GM). Within the excitation range from 770 to 860 nm, we determined φFϬ2 values of 7.1 GM at 800 nm and 7.7 GM at 810 nm for TPA-trisNTA (Fig. 2c), which were consistent with reported values of DEAC-caged compounds51. The fluorescence quantum yield φF of TPA-trisNTA was determined to be 15% (Supplementary Methods 1.5, Supplementary Fig. 7). To examine the 2P-activation, we tested the power dependent 2P absorption of TPA-trisNTA at 800 nm by varying the excitation energies from 20 to 100 mW (Fig. 2d). The double-logarithmical dependence of excitation energy versus fluorescence intensity of 1.93 ± 0.02 reflects an almost perfect quadratic power dependency (Fig. 2e) and confirms a 2P-activation process. These results demonstrate that TPA-trisNTA undergoes a fast one-photon photoscission and can be activated by two-photon excitation.
Photoinstructive matrices for 3D protein assembly
To advance 3D protein assembly, we engineered 2P-photoinstructive hydrogels. TPA-trisNTA was coupled to a maleimide polyvinyl alcohol (PVA) precursor (Supplementary Fig. 8) and immediately cross-linked with a dithiol polyethylene glycol (PEG). We investigated the global one-photon activation of TPA-trisNTA in hydrogels by generating large circular regions within the scaffold (Fig. 3a) using mask-patterning (405 nm LED lamp, 185 mW/cm2, 1 min). Visualization of the illuminated areas was realized by binding of GFP-His6 (300 nM) followed by confocal laser-scanning microscopy (CLSM) (Fig. 3b). GFP-His6 was exclusively assembled in photoactivated areas with excellent signal-to-noise ratio, highlighting the specificity of the trisNTA/His-tag interaction. Patterning occurred in 3D with a penetration depth of 100 µm into the scaffold. Although mask lithography offers only limited spatial resolution, the rapid structuring of large areas with well-defined bulk hydrogel properties can be realized. In contrast, laser lithography allows to photostructure hydrogels with high spatial flexibility over protein assembly (Fig. 3c). We therefore exploited laser-scanning activation of TPA-trisNTA in a spatially defined region-of-interest (ROI: heart) using either UV light (405 nm) or focused NIR fs-laser pulses at 800 nm. ROI visualization was followed by binding of a fluorescent His6-peptide (His6-AF647). In both cases, a localized photoreaction proximal to the focal point in x/y direction was observed. In the focal plane, however, a well-defined photoactivation and thus protein assembly was received for 2P-patterning compared to UV activation. Analyzing the protein patterning in z direction illustrated the improved 3D resolution for the photoreaction. Via 2P-activation processes, a localized and precise activation of TPA-trisNTA and thus POI alignment in z direction was accomplished, which is proven by a sharp image in the focal plane and a z resolution of ~10 µm. In contrast, conventional laser-scanning lithography resulted in a conical shape of protein alignment and a z resolution >40 µm. Owing to photoreactions initiated above and below the focal plane, partial protein assembly throughout the beam path is induced by 1P activation. This verifies that focused 2P-activation of TPA-trisNTA allows the accurate design of volumetric structures in hydrogels in 3D.

a Schematic illustration of photoactivation in hydrogels by mask patterning or laser lithography using UV or NIR light. b Mask patterning of circular structures using a 405 nm UV/LED system (185 mW/cm2 for 60 s) showed a very good contrast between illuminated and non-illuminated areas (top). Photopatterning occurred in three dimensions with a penetration depth of 100 µm and uniform organization of GFP-His6 (bottom). 3D structures were visualized by immobilization of GFP-His6, and images were reconstructed from a series of z stacks. Scale bars: 100 µm. c In-situ laser lithography of TPA-trisNTA and assembly of AF647-labeled His-tagged POI within heart-shaped ROIs via one-photon excitation at 405 nm (4.5 mW) or two-photon excitation at 800 nm (5.5 mW). One-photon activation occurred primarily proximal to the focal plane with vicinal photoactivation of TPA-trisNTA above and below the focal plane, resulting in a conical shape of POI alignment in the gel. Two-photon excitation of TPA-trisNTA led to a sharp and precise POI assembly in x/y direction and localized activation in z dimension, accentuating the superior control over photoactivation and subsequently over protein organization. 3D images were reconstructed after collecting a series of z stacks by CLSM recording. Scale bars: 20 µm.
In-gel photoevolution of TPA-trisNTA
The precise 2P-patterning of proteins within hydrogels and the very sensitive TPA-trisNTA photochemistry was further explored in terms of wavelength-selective activation and resolution in 3D. To examine the in-gel excitation profile of TPA-trisNTA, we tested activation wavelengths ranging from 720 to 800 nm with 20 nm increments and a constant laser power of 5 mW (Fig. 4a). After binding of His6-AF647, the fluorescence intensities of the activated ROIs were visualized by CLSM. Assessed by an intensity profile through the focal plane, the photoscission of TPA-trisNTA increases gradually to the wavelength increment with the strongest TPA-trisNTA photoactivation at 800 nm. This result is in good agreement with the 2P absorption profile determined in solution (Fig. 2c).

a TPA-trisNTA functionalized hydrogel was activated with wavelengths from λex = 720–800 nm at 5 mW. Intensity profiles through the sum of z slices of immobilized AF647-POI showed the best photoactivation at 800 nm. For technical reasons, wavelengths higher than 800 nm are not applicable. b Assignment of the size limit for photostructuring in x/y dimension by writing six quadratic ROIs of decreasing size (50, 25, 12, 6, 3, and 2 µm) into the gel. Intensity profiles of the ROIs demonstrated constant fluorescence intensities heights in all ROIs except for the 2 µm-ROI. c Determination of the resolution limit via two-photon structuring of four individual diffraction-limited squares in x/y direction. Spots separated by 1 µm or more can be clearly distinguished by CLSM imaging, which is also reflected in the intensity profile. Scale bar: 5 µm. d 3D POI network constructed by two-photon activation of TPA-trisNTA. Three circular spots were written in different z layers to investigate the z resolution. Intensity profiles of AF647-POI in z direction showed spatially localized photoactivation with clearly distinguishable edges of the three written ROIs. a.u. corresponds to arbitrary units. Scale bar: 50 µm.
The smallest scale of possible features in ROIs relies on the resolution of the scanning process. To determine the minimal patterning area in x/y dimension, we wrote six quadratic ROIs of decreasing size (50, 25, 12, 6, 3, and 2 µm) by utilizing 800 nm for TPA-trisNTA photoactivation (Fig. 4b). Photopatterns were again visualized via immobilization of His6-AF647 followed by CLSM. Even the 2 µm ROI could precisely be written into the hydrogel. However, the intensity profile of the ROIs revealed a diminished fluorescence intensity of the 2 µm ROI compared to the 3 µm or larger ROIs. Under these 2P-patterning conditions, the minimal ROI size is between 2 and 3 µm and can be locally specified with single-digit micrometer precision in the hydrogel.
We next determined the x/y resolution of the writing process and thus the ability of our approach to discern individual diffraction-limited spots. Therefore, varying sets of four adjacent quadratic ROIs (5 µm) were written into the hydrogel with successively decreased spacing between the squares (Fig. 4c). Whereas photopatterns with an inter-ROI distance of 1 µm or larger were clearly distinguishable, shorter ROI-spacing resulted in fuzzy and barely discriminable photopatterned areas. The shorter-spaced ROIs could no longer be spatially classified into individual patterns, indicating that an inter-ROI distance of 1 µm or larger is required for accurate photopatterning.
The major advantage of 2P-excitation is the precise control of activation in z dimension. To demonstrate a defined 3D protein organization, circular ROIs at different z locations and 10 µm distance to each other, were photostructured into the TPA-trisNTA-functionalized hydrogel (Fig. 4d). In x/y- as well as in z dimension a homogenous and spatially defined protein arrangement was detected by CLSM. The intensity profiles along the z axis showed distinct and clearly distinguishable photopatterned regions, further highlighting the 3D precision of the 2P-writing process. A z resolution of 5 ± 1 µm (FWHM) was determined for the 2P-activation process, which is in good agreement with typically observed z resolutions. A major difference of the trisNTA-His-tag system compared to other covalent 2P-patterning strategies is its reversibility42. For orthogonal detachment of the POI, the hydrogel was thus exposed to 500 mM imidazole. CLSM imaging before and after treatment revealed a rapid release of the immobilized His-tagged POI (Supplementary Methods 1.6, Supplementary Fig. 9). Pattern recovery was readily facilitated by immobilization of another His-tagged POI, permitting write-read-erase applications as well as enable repetitive cycles of protein patterning and release. We also assessed the long-term stability and persistence of the photopatterns. Remarkably, imaging the written ROIs ten days after 2P-structuring showed that the gel was still decorated with the POI. After this prolonged time, a fivefold decreased fluorescence intensity was observed (Supplementary Methods 1.7, Supplementary Fig. 10). All images were taken in HBS buffer pH 7.2 at 25 °C. On day 10, fresh HBS buffer pH 7.2 was added before imaging the ROI. Important to note, cell culture media may affect the kinetic complex stability and thus critically reduce the long-term stability of the trisNTA/His-tag interaction, particularly when histidine-rich buffers will be utilized. Overall, the modularity and reversibility of TPA-trisNTA offers iterative tethering and replacement of proteins as well as regeneration of hydrogels.
Photosensitizers amplify the two-photon patterning
Encouraged by these results, we aimed to improve the photoactivation efficacy and sensitivity of TPA-trisNTA under 2P-excitation. By acting as auxiliary chromophore, 2P active fluorophores efficiently absorb NIR light and transfer the absorbed energy to acceptor photocages to enhance photoscission. Although a covalent tandem system for intramolecular energy transfer is more efficient and provides an acceptor-donor ratio of 1:1, we strived for a bimolecular sensitization through diffusive encounter52,53. Hence, we investigated the effect of the 2P active fluorophores ATTO390 and rhodamine B (RB) during the 2P-patterning process. Both fluorophores have large 2P absorption cross sections (ATTO390: 14 GM; RB: 150 GM) and are fully biocompatible, which is important for cell applications53,54. By writing a matrix of 20 quadratic ROIs in the hydrogel, we systematically modified the light dose of the 800 nm laser (0.5–5.5 mW or 0.3–2.2% laser power versus 6–30 s illumination time) (Fig. 5a). For quantification, the fluorescence intensities emanating from the entire activated volume (voxel) of each ROI were analyzed. During writing, the TPA-trisNTA functionalized hydrogels were exposed to distinct concentrations of ATTO390 (50, 100, and 500 µM) or RB (50 and 100 µM) or buffer. In total, 120 conditions were screened to systematically optimize the 2P accessibility of our optochemical tool as well as to establish robust protocols for arbitrary 2P protein patterning (Supplementary Fig. 11, Supplementary Fig. 12). For hydrogels immersed to HBS buffer, POI patterns were faintly visible below 3.1 mW at 18 s or 1.5 mW at 24 s, indicating that the photoscission of TPA-trisNTA occurred only to a minor degree. In contrast, hydrogels exposed to 100 µM ATTO390 or 50 µM RB and subjected to 2P-patterning showed a globally enhanced POI recruitment and thus 2P-activation of the optochemical tool (Fig. 5a, Supplementary Fig. 13, Supplementary Fig. 14). In case of 50 µM RB, the onset of visibly detectable POI tethering was already displayed at a laser power of 0.5 mW at 18 s, whereas 1.5 mW at 6 s were required for 100 µM ATTO390. Generally, the presence of 50 µM RB induced a more efficient photoscission of TPA-trisNTA and thus enhanced 3D protein binding, which was expressed by increased fluorescence intensities as well as improved signal-to-noise ratio. For RB, even the lowest applied laser power effectually fostered protein pattering. In contrast, photostructuring in presence of 50 µM ATTO390 resulted in no significant increase in POI tethering and thus photoactivation of TPA-trisNTA. At higher ATTO390 or RB concentration, quenching effects of the auxiliary chromophore need to be considered and can thus stall an efficient energy transfer to TPA-trisNTA. Moreover, at high light doses (5.5 mW; 30 s), a halo-effect exceeding the ROI boundary was observed, concomitant with a diminished fluorescence intensity within the spot compared to surrounding ROIs. This observation is indicative for a destruction of the hydrogel in the focal plane. Indeed, a dumbbell-shaped pattern was visible in the z stack of the CLSM image with depleted POI tethering in the focal plane (Fig. 5b). Depending on the photoactivation conditions, extended photopatterned areas in z direction were visible with out of focus and diffuse protein tethering. The deteriorated POI recruitment is most likely a result of plasma-mediated ablation in combination with enhanced light scattering, also corroborated by the intensity profiles along the z axis (Supplementary Methods 1.8, Supplementary Fig. 15)55,56. Destructive effects were further promoted in 2P-patterned hydrogels immersed to higher 2P fluorophore concentrations. Overall, hydrogels photostructured in the presence of 100 µM ATTO390 and 50 µM RB under 3.1 mW at 12 s showed boosted TPA-trisNTA photoactivation without significant photodestruction. Compared to untreated gels, TPA-trisNTA activation was enhanced 3.8-fold for ATTO390 and 15.6-fold for RB (Fig. 5c) with these settings. As a corollary, the laser power can be reduced up to tenfold (5.5–0.5 mW) during protein alignment to obtain efficient protein densities. In our endeavor to improve the photoactivation efficacy, the 2P fluorophore sensitized photorelease of TPA-trisNTA simply advanced the 2P-writing process.

a TPA-trisNTA functionalized hydrogels were activated at 800 nm in the presence of HBS buffer, 100 µM ATTO390 or 50 µM RB with varying laser power and illumination time. With increased laser power or illumination time, an enhanced tethering of AF647-POI was observed, reporting on improved photoactivation of TPA-trisNTA. The presence of photosensitizers induced an intensified photoscission and consequently enriched POI binding. Scale bar: 15 µm. b Orthogonal cross section (x/z direction) of ATTO390 soaked hydrogel and photoactivated with increasing laser power for 12 s. With elevated laser power, an expanded z depth of the activated area was observed, finally ending up in a dumbbell-shaped pattern due to plasma-mediated ablation. c Comparison the fluorescence intensity of ROIs illuminated with 3.1 mW and 12 s demonstrated enhanced photoactivation of the gels exposed to photosensitizers. For 100 µM ATTO390, a 3.8-fold higher fluorescence intensity was detected compared to HBS buffer, and for a hydrogel photopatterned in presence of 50 µM RB, a 15.6-fold increased fluorescence intensity was determined (n = 3). a.u. corresponds to arbitrary units.
The high spatial positioning of biomolecules provided by light actuation constitutes photoinstructive hydrogels as powerful platforms for cellular applications. Hence, we tested the applicability of the TPA-trisNTA functionalized hydrogels for cell studies as well as the cytotoxicity of the 2P-sensitizers under illumination conditions. To evaluate cell viability in gels, living HeLa cells were embedded within the TPA-trisNTA functionalized scaffold to form a cell-laden hydrogel. The encapsulated HeLa cells were assessed by annexin V staining to report on apoptotic cells (Supplementary Fig. 16). A very high cell viability (91 ± 1%) was observed, confirming that cell embedment in the photoinstructive gel is non-toxic. Of note, Ni(II) ions complexed to trisNTA at concentrations up to 5 µM exhibit no cytotoxic effects on cells46. To estimate the cytotoxicity of the 2P-sensitizers under illumination conditions, cells were exposed to increasing concentrations of either ATTO390 or RB and analyzed via a 3-(4,5-dimethylthiazol-2yl)-2,5-dipehnyltetrazolium bromide (MTT) assay (Supplementary Methods 1.12, Supplementary Fig. 17). Both enhancers showed no dark toxicity up to 100 µM (ATTO390: > 70% cell viability; RB: > 90% cell viability). However, 500 µM ATTO390 severely affected the cell viability, both in the non-illuminated and illuminated state. In contrast, 500 µM RB hardly affected the cells at all (>70% cell viability). Hence, both sensitizers were cytocompatible at the optimized concentrations for laser lithography in the dark and particularly under light actuation. Even though these cell assays only partly demonstrate cell viability, we next aimed to prove general cell applicability of our photoinstructive matrices. First, mask patterning as well as in-situ lithography by one-photon illumination in presence of HeLa cells was performed. After mask patterning (405 nm LED lamp, 185 mW/cm2, 1 min) of a cell-laden hydrogel, tethering of His6-mCherry (300 nM) to distinct patterns and vicinity to living cells was realized. Visualization via CLSM showed the structuring of large areas with His6-POIs covering multiple cells (Supplementary Fig. 18). In addition, in-situ one-photon activation of a single cell stably expressing the G-protein coupled receptor Y2 equipped with an external His6-tag as well as an autofluorescent protein (His6-Y2RmEGFP) was done after embedding in a photoinstructive matrix. A defined ROI (10 × 20 µm) covering a cell hemisphere was illuminated with 405 nm at 4.5 mW/cm2 laser power and 200 s. The intensity profile of the GFP signal emanating from the Y2 receptor before and after illumination, illustrated the enrichment of the receptor to the light-exposed area and thus trisNTA mediated receptor assembly after photoactivation (Supplementary Fig. 19). Lastly, in-situ 2P-activation of a TPA-trisNTA functionalized hydrogel was performed in presence of live, intact HeLa cells. Various sized ROIs in vicinity to distinct single cells were simultaneously photo-patterned by 2P-scission of the optochemical tool at 800 nm (3.1 mW; 12 s) and subsequently decorated with His6-mCherry (300 nM). The precise, subcellular protein deposition at gel subvolumes in close proximity to living cells was accomplished, highlighting the applicability of the 2P-writing process via TPA-trisNTA in presence of live cells (Fig. 6, Supplementary Fig. 20). Here, we employed HeLa cells, which posses enhanced fitness and robustness toward external stimuli and are a widely used basic model for cell applications. Within the time frame of experiments (12 h), we did not observe any abnormal behavior of the cells after 2P-patterning, indicating basic cell compatibility. Since the studies were performed on single-cell level, they do not allow drawing conclusions of ensemble cell behavior yet. Although future studies are required to investigate long-term cell viability as well as to demonstrate the proof-of-principle with stem cells as model cell line, we believe that the results represent a valuable demonstration of the user-defined, spatially controlled protein patterning in cell-laden TPA-trisNTA functionalized hydrogels.

In close proximity to encapsulated HeLa cells stably expressing Y2RmEGFP, distinct ROIs (magenta boxes) were simultaneously photoactivated at 800 nm (3.1 mW, 18 s) and subsequently assembled with His6-mCherry (300 nM). Within the user-specified gel subvolumes, the 2P-activation of TPA-trisNTA created anchoring sites for the His6-POI in the photoinstructive hydrogel, demonstrating successful 2P-scisision of TPA-trisNTA in presence of and close proximity to living cells. Scale bar: 10 µm.
3D protein networks with single-digit micrometer precision
Finally, we aimed to transfer the enhanced 2P-writing process to small-scale protein alignment at selected loci in hydrogels. Typically, 2P lithography is applied to write 3D patterns on the order of tens of microns (>100 µm). While this scale is powerful for manipulating the behavior of larger cell networks or multicellular clusters, the ability of 2P-processes to in-situ modify hydrogels with micrometer-scale precision is underrepresented. To scrutinize TPA-trisNTA functionalized hydrogels on the order of a few microns in 3D, we created custom-made, spatially defined ROIs. Patterning was performed in the presence of 50 µM RB at 800 nm (3.1 mW; 12 s). Upon locally defined photoactivation of the portrait of Maria Goeppert-Mayer in three distinct z planes, fluorescently labeled POI was site-selectively immobilized (Fig. 7). Remarkably, the enhanced 2P-activation facilitated the writing of complex 3D patterns, exhibiting feature sizes with micrometer precision (cheek of Maria Goeppert-Mayer: ~ 1 µm) (Supplementary Fig. 21). The very good signal-to-background ratio between patterned and non-patterned areas is a result of the improved 2P lithography as well as high-affinity, site-selective binding of photoliberated trisNTA to His-tagged POI. With regard to fineness and precision of protein assembly in 3D, the TPA-trisNTA system extends the level of protein patterning as well as expands the repertoire of tools for protein immobilization. Noteworthy, the images were not background corrected and depict the genuine, current state. Important to mention, the apparent resolution of the imaging process is reduced compared to the precision of the writing process, since the observed resolution is a convolution of both processes. The microheterogeneity of the fluorescent signal in the different x/y planes depends on the efficiency of the 2P-uncaging process, which in turn is predetermined by the density of tethered TPA-trisNTA. In z dimension, a continuous decrease of the POI density with increasing z depth to ~40–70 µm was observed. Important to note, the penetration depth depends on both the laser intensity and the wavelength. Here, structuring was performed with a tenfold lower laser intensity compared to common 2P laser lithography approaches57. If required, additional depth can be achieved by increasing the laser power. Nevertheless, the 3D POI network of Maria Goeppert-Mayer clearly shows the power of 2P lithography to pattern freely designed 3D structures at small-scale and with good z resolution.

a The portrait of Maria Goeppert-Mayer was printed by two-photon excitation (800 nm, 3.1 mW, 12 s; presence of 50 µM RB) in three different z layers. Structures were visualized by assembly of AF647-POI followed by CLSM. The custom-made ROI design for laser activation is shown on the top right. b Orthogonal cross sections (x/z plane) of the two-photon patterned ROIs imaged by CLSM. Images displayed a sharp, spatially localized activation of TPA-trisNTA at µm-scale within multiple z layers and showed a confined immobilization of AF647-POI in z direction. c–e CLSM images show the respective focal plane (x,y) indicated by gray dashed lines. Scale bars: 10 µm.
Conclusion
In summary, by incorporation of TPA-trisNTA into hydrogels, we engineered a photoinstructive matrix, which was successively photostructured by 2P laser lithography with various His-tagged POIs. NIR-light guided protein organization offered an exquisite control of protein alignment in 3D with submicron resolution, culminating in well-resolved structures particularly along the z coordinate. By facile addition of 2P-sensitizers (ATTO390 or RB) as diffuse encounter, we devised a proceeding to significantly improve the efficiency of 2P laser lithography. This approach contributed to considerably reduce the laser power for 2P lithography and was successfully applied to photopattern custom designed volumetric structures at small-scale in 3D. As the engineered photoinstructive matrix showed cell applicability, our approach will be extremely useful for the arrangement of more complex bioactive proteins into matrices by NIR-light. Since methods for hydrogel photopatterning of fragile full-length proteins are cumbersome and thus rare, the highly localized protein tethering via trisNTA/His-tag interaction opens up new perspectives for a reliable and flexible protein patterning within hydrogels. As an exciting application, we currently aim to confine membrane-associated receptors in 3D as well as to assemble signaling molecules at defined subvolumes around hydrogel-embedded cells, multicellular clusters or organoids.
Methods
Synthesis of TPA-trisNTA
Fmoc-protected DEAC β-amino acid was synthesized in six steps and characterized by nuclear magnetic resonance (NMR) spectroscopy and high-resolution electrospray ionization mass spectrometry (HR ESI-MS). TPA-trisNTA (trisNTA-AC-DEAC-GHHHHH-OH) was synthesized through microwave-assisted Fmoc solid-phase methodology using the Liberty Blue microwave peptide synthesizer (CEM, Germany) and starting from a preloaded Fmoc-His(Trt)-Wang (Sigma Aldrich, Germany) resin. All reactions except for coupling of the Fmoc-protected DEAC β-amino acid (single coupling) were done twice with 0.2 M of Fmoc-protected amino acid, 0.5 M N,N’-diisopropylcarbodiimid, 0.5 M 1-hydroxybenzotriazole monohydrate in dimethylformamide (DMF). Fmoc-deprotection was performed with 20% (v/v) piperidine in DMF. OtBu-protected carboxy-trisNTA was manually coupled on the N-terminal glycine using (1-cyano-2-ethoxy-2-oxoethylidenamino-oxy)dimethylamino-morpholino-carbenium-hexafluoro-phosphat (COMU) and N,N-diisopropylethylamine (DIPEA) in DMF. Peptide cleavage/deprotection was achieved using 95% trifluoroacetic acid (TFA), 2.5% H2O and 2.5% 1,2-ethanedithiol followed by two precipitation cycles (50 mL diethyl ether, 0 °C). The crude peptide was purified via preparative reverse-phase high-performance liquid chromatography (RP-C18-HPLC) using a 30 min gradient (5–70% of acetonitrile and 0.1% TFA in H2O) and lyophilized (Supplementary Methods 1.3, Supplementary Fig. 4). TPA-trisNTA purity was confirmed by HR ESI-MS. The complete synthetic details and characterization of TPA-trisNTA and Fmoc-protected DEAC β-amino acid are given in the Supplementary Methods. For NMR and ESI spectra see Supplementary Figs. 22–36.
Photolysis reaction of the DEAC ß-amino acid in solution
50 µL aliquots of a stock solution of Fmoc-protected DEAC β-amino acid in HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl pH 7.2) were illuminated at 405 nm (185 mW/cm2, Thorlabs LED system). To study the kinetics of photolysis, samples were taken at different time points (0, 5, 10, 15, 30, 45, 60, 300, and 900 s). Photocleavage of Fmoc-protected DEAC β-amino acid was assessed by analytical RP-C18-HPLC (20 min gradient, 5–100% of acetonitrile and 0.1% TFA in H2O, Supplementary Methods 1.3, Supplementary Table 2), revealing conversion to uncaged DEAC β-amino acid with a different retention time (Supplementary Fig. 6). Photolysis efficiency was determined by evaluating the integral of the Fmoc-protected DEAC β-amino signal (tR = 17.0 min) as well as of the uncaged product (tR = 12.0 min). The photolysis kinetics were fitted with a mono-exponential function to determine the lifetime t1/2, indicating a fast one-step photoconversion.
Two-photon absorption spectrum of TPA-trisNTA
Two-photon excitation fluorescence (TPEF) measurements were performed using a tunable Ti:Sa laser (Tsunami, Spectra-Physics, USA) with a pulse duration of 150 fs and a 80 MHz repetition rate. The TPEF-signal was coupled into a spectrograph (SpectraPro 300i, Acton Research Corp., USA), equipped with a CCD-camera (EEV 400_1340F, Roper Scientific, USA). The excitation was adjusted to an average energy of 100 mW and the pulses were tightly focused on the sample compartment. The concentrations of the TPA-trisNTA and coumarin 307 were adjusted to 100 µM in a final volume of 300 µL. Coumarin 307 was used as reference, and values for calculation were taken from Xu et al.54. To obtain the two-photon absorption spectrum, the two-photon absorption action cross sections in the range of 770–870 nm were determined. After baseline and detector correction of the fluorescence spectra, the integrals were computed to obtain IF(X) and IF(R). For calculations the following equation was used:
with σ2 = two-photon absorption cross section, ϕF = fluorescence quantum yield, X = TPA-trisNTA, R = coumarin 307, IF = fluorescence intensity, c = concentration, η = refractive index of the solvent. The one-photon fluorescence quantum yield was assumed to equal the two-photon fluorescence quantum yield. The refractive indices of the samples (at 20 °C, Vis-NIR range) were determined to be η(R) = 1.33 (in MeOH) and η(X) = 1.33 (in H2O).
Two-photon power dependency measurement of TPA-trisNTA
To validate the two-photon absorption of TPA-trisNTA, a scan of the fluorescence intensity over a series of excitation energies (20–100 mW) was performed at a specific wavelength (800 nm). The count rate of the fluorescence response was logarithmically plotted against the logarithm of the excitation energy. A slope of 1.93 ± 0.02 was obtained, implicating an almost quadratic power dependency of the two-photon response. According to Bradley et al. deviations from a strict quadratic power dependency can occur due to heating effects or competing non-linear processes58.
Confocal imaging
Imaging was performed using a confocal laser-scanning microscope (LSM 880 AxioObserver, Carl Zeiss Microscopy, Germany), and images were taken with the Plan-Apochromat 20x (NA 0.8) and/or the Plan-Apochromat 63x/Oil (NA 1.4) objective. The following laser lines were used for excitation: 405 nm (diode laser) for Hoechst33342; 488 nm (argon laser) for green fluorescent protein (GFP-His6); and 633 nm (helium-neon laser) for His6-AF647 and Annexing V-AF647. All probes were imaged in HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C) or in case of live cells incorporated in the gel, the hydrogel was covered with Live Cell Imaging Solution (Invitrogen). The imaging process was controlled with the Zeiss Zen Black software. Image processing and evaluation was done with Fiji59.
Formation of TPA-trisNTA functionalized hydrogels
Gridded ibidi imaging dishes (µ-Dish 35 mm, high Grid-500, ibidi, Germany) were used for hydrogel preparation. For protein photopatterning, hydrogels were prepared from a commercial slow gelling 3-D Life PVA-PEG Hydrogel Kit (Cellendes, Germany). Gel formation was performed by mixing 0.6 µL HBS buffer pH 7.2 with 0.98 µL H2O and 1.20 µL maleimide-PVA (Supplementary Fig. 8). Then, 1.52 µL of a 790 µM TPA-trisNTA solution (aq.) was added and immediately mixed via pipetting and incubated for 5 min at RT in the dark. After a quick spin down, the reaction mixture was added to 1.74 µL dithiol-linker placed in the middle of an ibidi imaging dish and quickly mixed by pipetting. The network formation was allowed to proceed for 10 min in the dark. The solidified hydrogel was equilibrated with 1 mL HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C).
Laser-assisted photopatterning in hydrogels
For photostructuring by one- or two-photon excitation, a confocal laser-scanning microscope (LSM 880 AxioObserver, Carl Zeiss Microscopy, Germany) equipped with a Plan-Apochromat 63x/Oil (NA 1.4) objective, a 405 nm LED diode, and an ultrafast Ti:Sa Chameleon laser (Coherent Inc., Santa Clara, USA) with a pulse duration of 140 fs, a 80 MHz repetition rate and maximal output of 2.5 W tuned to 800 nm was employed for localized illumination. The user-defined ROI scanning mode and bleaching option were used to precisely control arbitrary patterns in 3D. The laser output power of the Ti:Sa Chameleon laser was precisely adjusted before each experiment to match 0.5, 1.5, 3.1 and 5.5 mW (corresponding to 0.3–0.7–1.2–2.2% of maximal laser power). The illumination time was 6, 12, 18, 24, or 30 s, equaling the sum of 500–2500 iterations. The scan speed was fixed at 1.54 μs/pixel. Photoactivation of hydrogels was performed in HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C). The illumination process was controlled with the Zeiss Zen Black software.
TPA-trisNTA mediated POI immobilization to hydrogels in 3D
Laser-treated hydrogels were washed five times with 2 mL HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C) prior to His-tagged POI tethering. All steps were performed at room temperature and protected from light. For POI patterning, the gel was covered with 1 mL of 10 mM NiCl2 in MQ water and incubated for 30 min followed by five times washing with HBS buffer (2 mL). Visualization of written structures was realized by incubation with 1 mL of 300 nM His6-GFP in HBS buffer or 300 nM His6-AF647 (HHHHHHSGGGSGGG-CAF647-A-NH2) in HBS buffer. After 30 min, the hydrogel was washed with HBS buffer (50 mL) by gently shaking for 30 min. Finally, the hydrogel was covered with 1 mL HBS buffer and subsequently imaged via CLSM (LSM 880 AxioObserver, Carl Zeiss).
Global photolithography of hydrogels
For photopatterning of large areas, hydrogels were exposed to UV light (λ = 405 nm LED lamp, 185 mW/cm2, 1 min) through a quartz mask with various sized chrome patterns on top of the gel. After illumination, the hydrogels were washed five times with HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C). POI binding and patterning was performed with His6-GFP after incubation with 10 mM NiCl2 in MQ. POI assembly was visualized by CLSM (LSM 880 AxioObserver, Carl Zeiss).
Cell viability assessed in cell-laden hydrogels
To analyze cell viability throughout encapsulation in hydrogels, a live-cell annexin V staining was performed to report on apoptotic cells. HeLa Kyoto cells (1 × 106 cells/mL) were washed and suspended in 100 µL annexin V binding buffer (Invitrogen). After adding 25 µL of annexin V-AF647 conjugate (Invitrogen) and one drop of NucBlue Live Ready Probes (ThermoFisher), cells were incubated for 15 min at room temperature and then washed once again with annexin V binding buffer. For hydrogel encapsulation, 10 µL of the HeLa cell suspension were mixed with 3.0 µL HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C), 11.3 µL H2O, 2.5 µL maleimide-PVA, 0.8 µL of a 20 mM TPA-trisNTA solution (aq.) and 3.0 µL dithiol-linker (Supplementary Table 4). After mixing via pipetting, hydrogel network formation was allowed to proceed for 10 min, followed by covering with 1 mL Live Cell Imaging Solution (Invitrogen). Cell viability analysis was performed by CLSM (Supplementary Fig. 16). Hoechst33342 stained nuclei were excited with 405 nm diode laser and HeLa cells in an early apoptosis state were displayed via the AF647 signal of the annexin V-AF647 conjugate and excited with the 633 nm laser. 708.5 µm × 708.5 µm images were taken. The total amount of cells indicated by the Hoechst33342 signal and the number of AF647-stained cells were counted with Fiji multipoint tool59. Determining the ratio between Hoechst33342- and AF647-stained cells revealed that 91 ± 1% of the encapsulated cells were vital. All experiments were performed in triplicates and error bars indicate the s.d. For general cell culture maintenance, see Supplementary Methods 1.10.
Preparation of cell-laden hydrogels for photopatterning of proteins
Y2R expressing HeLa cells were detached with 0.05% trypsin/0.02% EDTA/PBS (GE Healthcare) 16 h post receptor induction, centrifuged (300 × g for 3 min) and solubilized in 200 µL Life Cell Imaging Solution (Invitrogen). For hydrogel formation, 10 µL of the HeLa cell suspension were mixed with 3.0 µL HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C), 11.3 µL H2O, 2.5 µL maleimide-PVA, 0.8 µL of a 20 mM TPA-trisNTA solution (aq.) and 3.0 µL dithiol-linker. After mixing via pipetting, hydrogel network formation was allowed to proceed for 10 min. Subsequently, the hydrogel was covered with 1 mL Life Cell Imaging Solution. For mask lithography, hydrogels were exposed to UV light (λ = 405 nm LED lamp, 185 mW/cm2, 1 min) through a quartz mask with lattice designed chrome patterns on top of the gel (Supplementary Fig. 18). Laser lithography by one- or two-photon excitation was conducted on a confocal laser-scanning microscope utilizing the 405 nm LED diode (maximum output of 4.5 mW) or the ultrafast Ti:Sa Chameleon laser (Coherent Inc., Santa Clara, USA) tuned to 800 nm and a Plan-Apochromat 63x/Oil (NA 1.4) objective in Life Cell Imaging Solution at 37 °C. The written ROIs were visualized by binding of His6-mCherry (300 nM) or via following His6-Y2RmEGFP assembly at the activated cell hemisphere (Supplementary Fig. 19, Supplementary Fig. 20).
Photoenhancement effect of two-photon sensitizers in hydrogels
Photopatterning by two-photon excitation was conducted on a confocal laser-scanning microscope utilizing the ultrafast Ti:Sa Chameleon laser (Coherent Inc., Santa Clara, USA) tuned to 800 nm and a Plan-Apochromat 63x/Oil (NA 1.4) objective. Before each experiment, the laser intensity of the Ti:Sa Chameleon laser was precisely adjusted to match the given laser powers. 20 rectangular ROIs (15 µm width) were written into the gel with stepwise increased laser power (0.5, 1.5, 3.1, and 5.5 mW) or illumination time (6, 12, 18, 24, 30 s). The scan speed was fixed at 1.54 μs/pixel. Precise control of the laser dosage assigned to each ROI allowed generating photopatterns of different densities. Photoactivation of hydrogels was performed in HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C), in presence of ATTO390 (50, 100, 500 µM in HBS buffer) or of RB (50, 100 µM in HBS buffer). After rinsing with HBS buffer, Ni(II)-loading (10 mM in MQ water) and washing with HBS buffer, His6-AF647 (300 nM) tethering was performed. Via CLSM, z stacks (35 slices, z step width 1 μm) of the written ROIs were recorded. Imaging conditions (laser power, detector amplification and pinhole) were kept constant and applied to all photopatterned hydrogels. After background subtraction, z slices of each ROI were summed up and quantified by determining the total integrated fluorescence density of each voxel (Supplementary Fig. 11, Supplementary Fig. 12). For comparison, the total integrated fluorescence densities obtained under equal conditions were averaged (Supplementary Fig. 13, Supplementary Fig. 14). Each experiment was performed in triplicate or quadruplicate, and error bars indicate the s.d. Quantification was performed using Zeiss Zen Black and Fiji software59.
Intensified 3D arbitrary protein organization at low laser power
For locally precise two-photon excitation in hydrogels, the Ti:Sa Chameleon laser tuned to 800 nm and a Plan-Apochromat 63x/Oil (NA 1.4) objective on the confocal laser-scanning microscope were employed. Photostructuring was conducted in HBS buffer (20 mM HEPES/NaOH, 150 mM NaCl, pH 7.2, 25 °C) supplemented with 50 µM rhodamine B. By the user-defined ROI scanning option in combination with the bleaching mode of the microscope software (Zeiss Zen Black), the diverse shapes and size regions of the portrait of Maria Goeppert-Mayer were precisely written in x/y/z direction. The different ROIs in each z plane were photoactivated for 12 s with an average energy of 3.1 mW (1.2%; max output 2.5 W), a scan speed of 1.54 µs/pixel and a z stack thickness of 5 μm separated by 30 μm. The written structures were visualized by washing with HBS buffer followed by Ni(II)-loading (10 mM NiCl2 in MQ) and subsequent POI binding via incubation with 300 nM His6-AF647 in HBS buffer at room temperature. The POI assembly was recorded by CLSM (118 slices z stacks, 96 µm).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. The raw data underlying any graph is provided as a source data file.
References
Lee, M., Rizzo, R., Surman, F. & Zenobi-Wong, M. Guiding lights: tissue bioprinting using photoactivated materials. Chem. Rev. 120, 10670–10747 (2020).
Ruskowitz, E. R. & DeForest, C. A. Photoresponsive biomaterials for targeted drug delivery and 4D cell culture. Nat. Rev. Mater. 3, 17087 (2018).
Unal, A. Z. & West, J. L. Synthetic ECM: Bioactive synthetic hydrogels for 3D tissue engineering. Bioconjugate Chem. 31, 2253–2271 (2020).
Tam, R. Y., Smith, L. J. & Shoichet, M. S. Engineering cellular microenvironments with photo- and enzymatically responsive hydrogels: Toward biomimetic 3D cell culture models. Accounts Chem. Res. 50, 703–713 (2017).
Zhang, Y. S. & Khademhosseini, A. Advances in engineering hydrogels. Science (New York, N.Y.) 356, eaaf3627 (2017).
Rosales, A. M. & Anseth, K. S. The design of reversible hydrogels to capture extracellular matrix dynamics. Nat. Rev. Mater. 1, 15012 (2016).
Burdick, J. A. & Murphy, W. L. Moving from static to dynamic complexity in hydrogel design. Nat. Commun. 3, 1269 (2012).
Matsumoto, S. et al. Photo-responsive gel droplet as a nano- or pico-litre container comprising a supramolecular hydrogel. Chem. Commun. 44, 1545–1547 (2008).
He, M. T., Li, J. B., Tan, S., Wang, R. Z. & Zhang, Y. Photodegradable supramolecular hydrogels with fluorescence turn-on reporter for photomodulation of cellular microenvironments. J. Am. Chem. Soc. 135, 18718–18721 (2013).
Li, X. M., Gao, Y., Kuang, Y. & Xu, B. Enzymatic formation of a photoresponsive supramolecular hydrogel. Chem. Commun. 46, 5364–5366 (2010).
McNitt, C. D., Cheng, H., Ullrich, S., Popik, V. V. & Bjerknes, M. Multiphoton activation of photo-strain-promoted azide alkyne cycloaddition “click” reagents enables in situ labeling with submicrometer resolution. J. Am Chem. Soc. 139, 14029–14032 (2017).
Azagarsamy, M. A. & Anseth, K. S. Wavelength-controlled photocleavage for the orthogonal and sequential release of multiple proteins. Angewandte Chemie-Int. Ed. 52, 13803–13807 (2013).
DeForest, C. A., Sims, E. A. & Anseth, K. S. Peptide-functionalized click hydrogels with independently tunable mechanics and chemical functionality for 3D cell culture. Chem. Mater. 22, 4783–4790 (2010).
DeForest, C. A. & Anseth, K. S. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem. 3, 925–931 (2011).
Adzima, B. J. et al. Spatial and temporal control of the alkyne-azide cycloaddition by photoinitiated Cu(II) reduction. Nat. Chem. 3, 256–259 (2011).
DeForest, C. A. & Anseth, K. S. Photoreversible patterning of biomolecules within click-based hydrogels. Angewandte Chemie-Int. Ed. 51, 1816–1819 (2012).
DeForest, C. A., Polizzotti, B. D. & Anseth, K. S. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater. 8, 659–664 (2009).
Luo, Y. & Shoichet, M. S. A photolabile hydrogel for guided three-dimensional cell growth and migration. Nat. Mater. 3, 249–253 (2004).
Griffin, D. R. & Kasko, A. M. Photodegradable macromers and hydrogels for live cell encapsulation and release. J. Am Chem. Soc. 134, 13103–13107 (2012).
Kloxin, A. M., Kasko, A. M., Salinas, C. N. & Anseth, K. S. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science (New York, N.Y.) 324, 59–63 (2009).
Wosnick, J. H. & Shoichet, M. S. Three-dimensional chemical patterning of transparent hydrogels. Chem. Mater. 20, 55–60 (2008).
Wylie, R. G. et al. Spatially controlled simultaneous patterning of multiple growth factors in three-dimensional hydrogels. Nat. Mater. 10, 799–806 (2011).
Seliktar, D. Designing cell-compatible hydrogels for biomedical applications. Science (New York, N.Y.) 336, 1124–1128 (2012).
DeForest, C. A. & Anseth, K. S. Advances in bioactive hydrogels to probe and direct cell fate. Ann. Rev. Chem. Biomol. Eng. Vol 3 3, 421–444 (2012).
LaFratta, C. N., Fourkas, J. T., Baldacchini, T. & Farrer, R. A. Multiphoton fabrication. Angewandte Chemie-Int. Ed. 46, 6238–6258 (2007).
Tibbitt, M. W., Shadish, J. A. & DeForest, C. A. Photopolymers for Multiphoton Lithography in Biomaterials and Hydrogels. In Multiphoton Lithography: Techniques, Materials, and Applications 183–220 (Wiley Publishing, 2016).
Wang, L. & Huang, X. in Artificial Protein and Peptide Nanofibers. (eds. G. Wei & S. G. Kumbar) 415–436 (Woodhead Publishing, 2020).
Gonçalves, A. et al. Trends in Protein-Based Biosensor Assemblies for Drug Screening and Pharmaceutical Kinetic Studies. Molecules (Basel, Switzerland) 19, 12461–12485 (2014).
Yuan, Y. et al. Conjugated polymer and drug co-encapsulated nanoparticles for Chemo- and Photo-thermal Combination Therapy with two-photon regulated fast drug release. Nanoscale 7, 3067–3076 (2015).
Mynar, J. L. et al. Two-photon degradable supramolecular assemblies of linear-dendritic copolymers. Chem. Commun. 43, 2081–2082 (2007).
Miyata, T., Asami, N. & Uragami, T. A reversibly antigen-responsive hydrogel. Nature 399, 766–769 (1999).
Ehrick, J. D. et al. Genetically engineered protein in hydrogels tailors stimuli-responsive characteristics. Nat. Mater. 4, 298–302 (2005).
Murphy, W. L., Dillmore, W. S., Modica, J. & Mrksich, M. Dynamic hydrogels: Translating a protein conformational change into macroscopic motion. Angewandte Chemie-Int. Ed. 46, 3066–3069 (2007).
Alge, D. L., Azagarsamy, M. A., Donohue, D. F. & Anseth, K. S. Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine-norbornene chemistry. Biomacromolecules 14, 949–953 (2013).
Hahn, M. S., Miller, J. S. & West, J. L. Three-dimensional biochemical and biomechanical patterning of hydrogels for guiding cell behavior. Adv. Mater. 18, 2679 (2006). +.
Fisher, S. A. et al. Photo-immobilized EGF chemical gradients differentially impact breast cancer cell invasion and drug response in defined 3D hydrogels. Biomaterials 178, 751–766 (2018).
DeForest, C. A. & Tirrell, D. A. A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat. Mater. 14, 523–531 (2015).
Mosiewicz, K. A. et al. In situ cell manipulation through enzymatic hydrogel photopatterning. Nat. Mater. 12, 1071–1077 (2013).
Shadish, J. A., Benuska, G. M. & DeForest, C. A. Bioactive site-specifically modified proteins for 4D patterning of gel biomaterials. Nat. Mater. 18, 1005–1014 (2019).
Broguiere, N. et al. Morphogenesis guided by 3D patterning of growth factors in biological matrices. Adv. Mater. 32, 1908299 (2020).
Grunwald, C. et al. In situ assembly of macromolecular complexes triggered by light. Proc. Natl Acad. Sci. USA 107, 6146–6151 (2010).
Labòria, N., Wieneke, R. & Tampé, R. Control of nanomolar interaction and in situ assembly of proteins in four dimensions by light. Angewandte Chemie-Int. Ed. 52, 848–853 (2013).
Gatterdam, K., Joest, E. F., Gatterdam, V. & Tampé, R. The Scaffold Design of Trivalent Chelator Heads Dictates Affinity and Stability for Labeling His-tagged Proteins in vitro and in Cells. Angewandte Chemie Int. Ed. 57, 12395–12399 (2018).
Wieneke, R. & Tampé, R. Multivalent chelators for in vivo protein labeling. Angewandte Chemie Int. Ed. 58, 8278–8290 (2019).
Lata, S., Reichel, A., Brock, R., Tampé, R. & Piehler, J. High-affinity adaptors for switchable recognition of histidine-tagged proteins. J. Am Chem. Soc. 127, 10205–10215 (2005).
Wieneke, R. et al. Live-cell targeting of His-tagged proteins by multivalent N-nitrilotriacetic acid carrier complexes. J. Am. Chem. Soc. 136, 13975–13978 (2014).
Kollmannsperger, A. et al. Live-cell protein labelling with nanometre precision by cell squeezing. Nat. Commun. 7, 10372 (2016).
Sánchez, M. F., Els-Heindl, S., Beck-Sickinger, A. G., Wieneke, R. & Tampé, R. Photoinduced receptor confinement drives ligand-independent GPCR signaling. Science (New York, N.Y.) 371, eabb7657 (2021).
Hagen, V. et al. Highly efficient and ultrafast phototriggers for cAMP and cGMP by using long-wavelength UV/Vis-activation. Angewandte Chemie Int. Ed. 40, 1045–1048 (2001).
LeValley, P. J. et al. Photolabile linkers: exploiting labile bond chemistry to control mode and rate of hydrogel degradation and protein release. J. Am Chem. Soc. 142, 4671–4679 (2020).
Goegan, B., Terzi, F., Bolze, F., Cambridge, S. & Specht, A. Synthesis and characterization of photoactivatable doxycycline analogues bearing two-photon-sensitive photoremovable groups suitable for light-induced gene expression. Chembiochem 19, 1341–1348 (2018).
Klausen, M., Dubois, V., Verlhac, J. B. & Blanchard-Desce, M. Tandem Systems for Two-Photon Uncaging of Bioactive Molecules. Chempluschem 84, 589–598 (2019).
Hammer, C. A. et al. Sensitized Two-Photon Activation of Coumarin Photocages. J. Phys. Chem. Lett. 9, 1448–1453 (2018).
Xu, C. & Webb, W. W. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J. Opt. Soc. Am. B 13, 481–491 (1996).
Pradhan, S., Keller, K. A., Sperduto, J. L. & Slater, J. H. Fundamentals of laser-based hydrogel degradation and applications in cell and tissue engineering. Adv. Healthcare Mater. 6, 1700681 (2017).
Tsai, P. S. et al. Plasma-mediated ablation: an optical tool for submicrometer surgery on neuronal and vascular systems. Curr. Opin. Biotechnol. 20, 90–99 (2009).
Lunzer, M. et al. A modular approach to sensitized two‐photon patterning of photodegradable hydrogels. Angewandte Chemie Int. Ed. 57, 15122–15127 (2018).
Bradley, D. J. et al. Interactions of picosecond laser pulses with organic molecules I. Two-photon fluorescence quenching and singlet states excitation in Rhodamine dyes. Proc. Royal Soc. London. A. Mathematical Phys. Sci. 328, 97–121 (1997).
Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Acknowledgements
This research was supported by the German Research Foundation (GRK 1986 to R.W. and R.T.), the LOEWE program (DynaMem A03 to R.W. and R.T.), the Volkswagen Foundation (Az. 96 498 to R.W. and Az. 96 496 to R.T.) and the Reinhart Koselleck Project (TA 157/12–1 to R.T.). The authors thank all members of the Institute of Biochemistry (Goethe University Frankfurt) for helpful discussion and comments. We thank Christian Winter for his support with the peptide synthesizer and LC-MS and Yagmur Aydogan for assistance with the TPEF measurements.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
H.K. performed the synthesis, experiments, and analysis. M.A. carried out the two-photon absorption measurements, the fluorescence quantum yield measurements and power-dependency measurements of TPA-trisNTA. J.W. was involved in analysis and discussion. H.K., R.T., and R.W. designed the experiments and analyzed the data. H.K. and R.W. wrote the paper. R.W. conceived the study and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Steven Caliari and John Plummer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krüger, H., Asido, M., Wachtveitl, J. et al. Sensitizer-enhanced two-photon patterning of biomolecules in photoinstructive hydrogels. Commun Mater 3, 9 (2022). https://doi.org/10.1038/s43246-022-00230-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-022-00230-w
- Springer Nature Limited
This article is cited by
-
Light-based vat-polymerization bioprinting
Nature Reviews Methods Primers (2023)