
Abstract
Microglia in hemorrhagic stroke contribute to both acute-phase exacerbation and late-phase attenuation of injury. Here, by using the mouse model, we reported that the shift in polarization of microglia from M1 to M2 phenotype could be altered by a past ‘mini’ stroke, resulting in better neurological function recovery, faster attenuation of lesion volume, and better survival. In mice with a previous stroke, M2 predominance appeared markedly in advance compared to mice without a previous stroke. Mechanistically, the RBC-mediated M2 polarization of microglia was synergistically enhanced by T cells: microglia cocultured with RBCs alone resulted in mild alterations to M2 markers, whereas in the presence of T cells, they expressed an early and sustained M2 response. These results suggest that by harnessing the microglia-mediated M2 polarization response, we could help mitigate devastating sequelae before a prospective hemorrhagic stroke even happens.
Similar content being viewed by others


Introduction
Hemorrhagic stroke (HS) refers to an intracerebral rupture of blood vessels leading to injury to the brain1. Spontaneous HS due to hypertension most commonly occurs in the putamen and caudate nucleus (50–60%)1. The overall prognosis is poor, with a 30-day mortality rate of 35–52%2. One-half of these deaths occur within the first two days2, stressing the importance of acute brain injury in pathogenesis. The mechanisms of injury within the first few hours include the hematoma mass effect and ensuing toxicity of red blood cell (RBC) components. The release of free hemoglobin, heme, and iron, followed by the hemolytic cascade, triggers a second wave of damage after the initial bleed3. As blood enters the normally blood-free brain parenchyma, reactive immune responses commence. The dormant immune cells within the brain, microglia, respond to injury within minutes to release chemokines and cytokines, propagating the inflammatory process and secondary brain insults4.
Microglia migrate caudally during embryonic development to stay within the brain parenchyma throughout life5. They are similar to bone marrow-derived macrophages morphologically and functionally, with the ability to phagocytose and present foreign antigens to T cells via major histocompatibility complex (MHC) class II molecules (MHC-II). Microglia, comprising 10–20% of the total cells within the brain, also surveil the cerebral immune environment and contribute to many major neuro-inflammatory actions of various diseases such as ischemic stroke, multiple sclerosis, traumatic brain injury, and infections6,7.
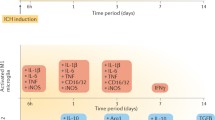
Following an HS, microglia first polarize via the classic polarization pathway, resulting in a phenotypic spectrum commonly deemed as M1 microglia8. This group is characterized by its pro-inflammatory actions that contribute to perilesional edema and exacerbate a cerebral injury. As time progresses, another polarization pathway, which results in a phenotype broadly classified as M2 microglia starts to assume dominance9. M2 microglia comprises a wide spectrum of cells that promotes cell regeneration (M2a), phagocytosis of hemolytic products (M2b and M2c), and removal of cell debris (M2b and M2c). This shift in polarization from M1 to M2 normally takes place on day 79. Although the dichotomization of microglia into either M1/M2 phenotype is overgeneralized, for the sake of simplicity we would refer to them as such within this study.
In the present study, we explored an incidental finding: mice that had undergone two separate episodes of HS performed significantly better on functional studies when compared with those without a previous HS. This phenomenon has never been discussed previously, and we aimed to determine the mechanisms underlying the above observation. Interestingly, we also observed the modulatory effects of a previous HS on subsequent strokes for the first time. Behavioral studies showed a significant trend, so we further performed MRI imaging, demonstrating attenuation of total lesion volume in animals with a previous HS. A recent study has put forth a concept of priming stimuli as mice that received multiple peripheral lipopolysaccharide injections showed attenuation of inflammation when exposed to Alzheimer’s disease and ischemic stroke10. As microglia are responsible for removing hematoma and cell debris7). In vitro studies revealed that microglia had retained innate immune memory during the previous stroke and that T cells were involved in this protective mechanism regarding earlier alternative microglia polarization. Our incidental discovery presented new evidence on a mechanism regarding the potential clinical impacts of successive hemorrhagic strokes which may pave the road to new, anticipatory therapeutic interventions that have never been seen before.

The first hemorrhagic stroke initiated a cascade of inflammatory actions downstream that corresponds to what others have observed in the past: M1 microglia take dominance initially before gradually declining to give way to an M2 predominant profile at day 7 post hemorrhagic stroke. Microglia then return to baseline levels. When a second hemorrhagic stroke was introduced, microglia no longer followed the former polarization profile but rather assumed an M2 predominant phenotype at an early stage. The group of mice with a previous stroke demonstrated improved survival rates, better functional outcomes, and rapid lesion resolution, compared to mice that were only encountering a hemorrhagic stroke for the first time.
Methods
Hemorrhagic stroke animal model
All animal experiments were approved by the Committee on the Use of Live Animals in Teaching and Research of our institution. Male C57 (C57BL/6 N, Charles River Lab, USA) mice 10–12 weeks old underwent all the described experiments unless otherwise specified (e.g., immune-deficient mice experiment design).
Mice were randomly divided into four groups. The sham group underwent two sham operations on days −14 and 0. The “past mini-stroke only” group underwent a single minor stroke on day −14. The “previous stroke group” underwent a minor HS at the left basal ganglia on day 14 and a major HS on the right on day 0. The control group received only one major HS at the right basal ganglia, with a sham operation on day −14. (Fig. 1a). For induction of the major HS, a replicable model was conducted as described before13. Mice were anesthetized via intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). A stereotactic device was used. Mice were secured in a prone position, and scalp fur was removed. A midline scalp single incision was made. Intracranial bleeding was induced by inserting a 26-gauge needle (10 uL Hamilton syringe) at 3.5 mm below the dura into the right basal ganglia at coordinates (x, y) = (+0.2 mm, +0.05 mm), with the bregma as (0,0), through a burr hole. One minute later, 0.1units of collagenase (0.25 U/µL, 0.4 µL) (Sigma-Aldrich, St. Louis, MI, United States) was injected at a rate of 0.1 µL/min with an infusion pump. After needle retraction, the burr hole was covered with bone wax, and the scalp was sutured. For a minor HS induction, the procedure was the same as mentioned above, with the only difference being that collagenase dosage was adjusted to 0.04 U (0.2 U/µL, 0.2 µL), and the injection site was at the left basal ganglia (−0.02 mm, +0.5 mm). Replicable hematomas were induced using this dosage and method for the minor HS (Supplementary Fig. 3). For a sham operation, the mice had a bur hole drilled at the same coordinates with needle insertion for 10 min before retraction.
In order to observe whether an absence of certain immune cells could interfere with functional recovery under the above study design, we used immune-modified mouse strains. Athymic nude mice (BALB/cAnN-nu, Charles River Lab, USA) and NOD-SCID mice (NOD.CB17-Prkdcscid/J, The Jackson Laboratory, USA) were used to repeat phenotypical experiments—similar sequences of HS for the “previous stroke group” and control group were performed on athymic nude mice, which lack mature T cells, and NOD-SCID mice, which lack mature B cells, T cell, and have defective NK cells.
Phenotypical observations
Mice were monitored on days 1, 3, 5, and 7 following the induction of a major hemorrhagic stroke. A blinded observer took the measurements.
Modified neurological severity score (mNSS)
The mNSS test, first described by Chen et al.36 and verified by validation studies37, quantifies motor, sensory, and balance deficits post-stroke. With a maximum score of 18 being the most severe, the gradual decline in neurological deficit scores traces the speed and extent of motor and sensory recovery after the initial injury37.
Rotarod
The Rotarod apparatus (AccuRotor, Omnitech electronics) rotates at an acceleration of 0.133 cm/s2. Results are recorded as latency-to-fall, which is the duration between commencement of rotation until the mouse falls off the rod36. Mice were first trained 7 days in three separate sessions prior to the major HS event. Subsequently, mice were assessed by the Rotarod test with the same acceleration profile. For each mouse, three runs were repeated, with 20-min rest intervals in-between.
Body weight
Mice were weighed individually every morning on days 1, 3, 5, and 7. Results were documented in grams.
Histopathologic examination
Mice were sacrificed on days 1, 3, 5, and 7 and perfused with 50 mL 4 °C normal saline. For the mini-stroke only and sham groups, mice were sacrificed 13, 9, 5, and 1 day before the major hemorrhagic stroke of other groups took place. Brains were sectioned into coronal slices of 2 mm thickness, and a photo was taken of each coronal section. Coronal images were imported to ImageJ, converted to grayscale, and a gradient-gated threshold was used to obtain the hematoma area. The sum of the sections from the cranial-caudal and caudal-cranial views were obtained, and the mean was multiplied by 2 mm (slice thickness) to obtain a final value of hematoma volume (expressed in mm3).
Magnetic resonance imaging (MRI) studies
A 7 T MRI scanner (PharmaScan 70/16, Bruder Biospin GmbH) was used to take 12 coronal plain scans, each of 0.5 mm thickness and positioned perpendicular to the axial and sagittal axes as previously described38. T2-weighted images were acquired as anatomical reference using a Rapid Acquisition with Refocused Echoes (RARE) sequence (FOV = 32 × 32 mm2, data matrix = 256 × 256, RARE factor = 8, TE/TR = 36/4200 ms). Images of the coronal plain were imported into ImageJ (version 1.8.0 for Windows, ImageJ Software). For each slice of the 12 coronal images, the total lesion area, defined as the hyperintense core, periphery, and surrounding rim39 was outlined manually. The sum of the combined lesion area from the 12 coronal slices was multiplied by slice thickness (0.5 mm) to obtain the total volume of the lesion. For statistical analysis, the total lesion volume was compared chronologically within the same mouse, with that on day 1 being presented as 100%. A subsequent decline in injury volume is expressed as a decrease in the percentage of residual volume compared to that on day 1.
Quantitative polymerase chain reaction (qPCR)
Mice were sacrificed 1, 3, 5, and 7 days after HS and perfused with 50 mL 4 °C normal saline. Brains were sectioned into slices of 2 mm thickness. Starting from the olfactory bulbs, the fourth and fifth slices were those with the largest hematoma area. These slices were harvested and subsequently divided into left and right sections. The right (injured) sections and enzymatically digested with RIPA buffer and PI. RNA was extracted from digested tissue, and cDNA was synthesized using a commercially available kit (Takara PrimeScript RT Reagent Kit). qRT-PCR using SYBR Green (QIAGEN) was performed to detect the subsequent genes: TNF-a, iNOS, CD206, and Arginine-1. The threshold cycle (CT) was normalized to GAPDH (ΔCT) of the same mouse and then to that of the sham mouse (ΔΔCT). Mean fold changes are depicted as 2(−ΔΔCT). Data acquisition was conducted in technical triplets. Primers are presented in Table 1.
Immunofluorescence staining
Mice were sacrificed and perfused with 50 mL cold PBS, followed by 50 mL 4% paraformaldehyde. Cryosections were obtained of the coronal plain at 5 μm thickness. Non-specific staining was blocked by 1% BSA for 1 h, followed by incubation with primary antibodies against microglia surface marker CD11b (1:100 Cell Signaling, Danvers, MA, USA), M2 marker CD163 (1:120 Cell Signaling) for 2 h at 4 °C. Slides were washed thrice before staining with secondary antibodies: anti-rabbit (Cell Signaling) and anti-mouse (Cell Signaling). DAPI nuclear staining (Sigma-Aldrich) was added to the mounting solution. Primary-cultured microglia were stained as follows: cells were fixed with 4% paraformaldehyde for 1 h, washed thrice with cold PBS, and stained with antibodies against Iba-1(1:100, Cell Signaling) for 2 h with nuclear staining DAPI (Sigma-Aldrich) added at the last 15 min. Cells were washed twice before incubation with secondary antibodies: anti-mouse (Cell Signaling) and anti-rabbit (Cell Signaling). All images were taken by the Invitrogen EVOS FL Color imaging system.
Flow cytometry
Mice from the previous stroke group and control group were sacrificed on days 1, 3, 5, and 7. Mice from the sham group and “past mini-stroke only” group were sacrificed on days −13, −9, −5, and −1. Peri-hematomal brain tissue was collected and mechanically homogenized by cutting with scissors, then digested by 5 mL 4% collagenase (Sigma-Aldrich, St. Louis, MI, United States) for 15 min at 37 °C. Digestion was halted by adding a 5 mL culture medium and single-cell suspension obtained by filtering through a 75 µm cell strainer. Cells were centrifuged at 400 × g for 5 min and resuspended in PBS. Cells were then washed again and resuspended in Flow Cytometry buffer (Cell signaling, Danvers, MA, USA) at a concentration of 105 cells per mL. For in vitro studies, microglia were scraped from the flask by a cell scraper at the experimental endpoint of each group. Cells were washed twice by centrifuging at 400 × g for 5 min and suspended in flow cytometry buffer. Each mL of suspended cells was incubated with conjugated flow antibodies (APC/Cy7 anti-mouse CD16/32 antibody, Biolegend #101327, 1:100; FITC anti-mouse CD206 antibody, Biolegend #141703, 1:80; Alexa Fluor 700 anti-mouse CD45.2 antibody, Biolegend #109822, 1:70; APC anti-mouse/human CD11b, Biolegend #101212, 1:100) for 1 h in darkness. For M2 verification, cells were additionally stained with PE anti-mouse CD163 antibody, Biolegend #156703. Cells were then washed twice by centrifuging at 400 × g for 5 min and resuspended in Flow Cytometry buffer. Analysis was conducted by CytoFLEX Flow Cytometer (Beckman Life Sciences, Indianapolis, IN, USA).
Microglia isolation and cell culture
In vitro experiments were conducted with primary-cultured microglia harvested from newborn pups of 0–3 days old, as previously described40. Pups were sacrificed by intraperitoneal injection of phenobarbital, then sprayed with 70% alcohol and decapitated with sterile equipment. The decapitated heads were rinsed in 4 °C PBS and immersed in dissection media. Under a microscope, the scalp was removed and the skull dissected along the midline up to the bregma, so that the brain could be scooped out using curving forceps. 5–6 brains were pooled and mechanically homogenized by cutting with scissors, then digested with 0.25% trypsin at 37 °C for 15 min. To end the digestion process, a 1.2 ml 1 mg/ml trypsin inhibitor was added and incubated for 1 min. The homogenized products were then centrifuged at 400 × g for 5 min and resuspended in warmed culture media (90% Dulbecco’s modified Eagle medium, Thermo Fisher Scientific, Gibco™; 10% fetal bovine serum, GE Healthcare, Hyclone™) twice, before being plated onto PDL coated T75 flasks at a density of 50,000 cells/cm2. Culture media was changed the next day, and subsequently every 5 days. This first step yields a confluent layer of astrocytes and microglia after 7 days.
To separate microglia from strongly adherent astrocytes, the flasks were shaken at 200 rpm for 2 h in a cell shaker maintained at 37 °C. The floating cells were collected and transferred to a newly coated T75 flask. This process yielded high quantity and high-quality microglia, which are ready for experimental use the next day.
Red blood cell (RBC) coculture
In order to mimic an HS in the cell culture setting, we added whole RBCs collected from the tail vein of mice at a ratio of 10:1 (RBC to microglia), as described before41. The coculture system was maintained at 37 °C for 1 h, and RBCs were subsequently removed via changing of cell medium. RBCs were non-adherent, while microglia were adherent to the flask wall.
Isolation of T cells
Splenic cells were obtained from adult C57 mice by homogenizing the whole spleen mechanically with scissors. RBCs were removed via lysis medium (Thermo Fisher, Waltham, MA, USA). Pan-naïve T cells were extracted via column-free magnetic cell isolation: first by positive selection (CD3 + CD45RA + CD197 + ), then negative selection (CD45RO−) using magnetic beads and a commercial kit (EasySep™ Human Naïve Pan T Cell Isolation Kit).
The collected pan-naïve T cells were added to Group F immediately after the first RBC coculture event (without direct contact with the RBCs) at a ratio of 1:1 (microglia to T cell). T cells were added to group C at the same time.
Erythrophagocytosis study
Whole RBCs were labeled with PKH-26 fluorescent probe (Sigma-Aldrich) and cocultured with primary microglia at a ratio of 10:1, as previously described33. After incubation at 37 °C for 1 h, the unengulfed RBCs were removed by washing thrice with PBS. Microglia were then harvested from the flask wall. The engulfed, intra-microglial RBCs were detected by flow cytometry via the FITC channel. The level of phagocytosis was quantified by the positive FITC percentage of each measurement.
Statistics and reproducibility
Phenotypical studies that include multiple measurements of the same mouse over time (body weight, mNSS, Rotarod, MRI total lesion volume) were analyzed by repeated-measurements two-way ANOVA with matched values of the same mouse across time (n = 10 for the previous stroke and control groups, n = 5 for the mini-stroke only and sham groups). Additionally, Sidak’s multiple comparisons test were conducted on the mean value obtained on each date between experimental groups (n = 25). Mechanistic studies that require sacrificing mice at the time of measurement (flow cytometry, qPCR) were analyzed by regular two-way ANOVA (no repeated measures) and Sidak’s multiple comparisons test (n = 7 for the previous stroke and control groups; n = 4 for the mini-stroke only and sham groups). Cell line studies involving more than two groups of importance to compare were analyzed by one-way ANOVA with Tukey’s multiple comparisons test. Cell line experiments were repeated three times within each group. The Gehan–Breslow–Wilcoxon test was used in the Kaplan–Meier survival curve analysis. A p value < 0.05 was considered statistically significant. GraphPad Prism version 6.04 for Windows, GraphPad Software was used.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data supporting the figures of this study is included in Supplementary Data 1.
References
Delcourt, C. et al. Intracerebral hemorrhage location and outcome among INTERACT2 participants. Neurology 88, 1408–1414 (2017).
Zia, E., Engström, G., Svensson, P. J., Norrving, B. & Pessah-Rasmussen, H. Three-year survival and stroke recurrence rates in patients with primary intracerebral hemorrhage. Stroke 40, 3567–3573 (2009).
Aronowski, J. & Hall, C. E. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurological Res. 27, 268–279 (2005).
Zhang, Z. et al. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol. Neurobiol. 54, 1874–1886 (2017).
Bachiller, S. et al. Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 12, 488 (2018).
Vay, S. U. et al. The plasticity of primary microglia and their multifaceted effects on endogenous neural stem cells in vitro and in vivo. J. Neuroinflammation 15, 226 (2018).
Taylor, R. A. & Sansing, L. H. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin. Dev. Immunol. 2013, 746068 (2013).
Tschoe, C., Bushnell, C. D., Duncan, P. W., Alexander-Miller, M. A. & Wolfe, S. Q. Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J. stroke 22, 29–46 (2020).
Lan, X., Han, X., Li, Q., Yang, Q. W. & Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 13, 420–433 (2017).
Wendeln, A. C. et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 (2018).
**, G., Keep, R. F. & Hoff, J. T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 5, 53–63 (2006).
Bai, Q., Xue, M. & Yong, V. W. Microglia and macrophage phenotypes in intracerebral haemorrhage injury: therapeutic opportunities. Brain 143, 1297–1314 (2020).
Taylor, R. A. et al. TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 127, 280–292 (2017).
Yang, J. et al. Interleukin-4 ameliorates the functional recovery of intracerebral hemorrhage through the alternative activation of microglia/macrophage. Front. Neurosci. 10, 61 (2016).
Hou, X. X., Wang, X. Q., Zhou, W. J. & Li, D. J. Regulatory T cells induce polarization of pro-repair macrophages by secreting sFGL2 into the endometriotic milieu. Commun. Biol. 4, 499 (2021).
Venkatasubramanian, C. et al. Natural history of perihematomal edema after intracerebral hemorrhage measured by serial magnetic resonance imaging. Stroke 42, 73–80 (2011).
Staykov, D. et al. Natural course of perihemorrhagic edema after intracerebral hemorrhage. Stroke 42, 2625–2629 (2011).
Li, N. et al. Temporal pattern of cytotoxic edema in the perihematomal region after intracerebral hemorrhage: a serial magnetic resonance imaging study. Stroke 44, 1144–1146 (2013).
Wang, J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 92, 463–477 (2010).
Kanazawa, M., Ninomiya, I., Hatakeyama, M., Takahashi, T. & Shimohata, T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18102135 (2017).
Liu, B. et al. CD163/hemoglobin oxygenase-1 pathway regulates inflammation in hematoma surrounding tissues after intracerebral hemorrhage. J. Stroke Cerebrovasc. Dis. 24, 2800–2809 (2015).
Lan, X. et al. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav. Immun. 61, 326–339 (2017).
Shalova, I. N. et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 42, 484–498 (2015).
Neher, J. J. & Cunningham, C. Priming microglia for innate immune memory in the brain. Trends Immunol. 40, 358–374 (2019).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Seong, S. Y. & Matzinger, P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 4, 469–478 (2004).
Netea, M. G., Latz, E., Mills, K. H. & O’Neill, L. A. Innate immune memory: a paradigm shift in understanding host defense. Nat. Immunol. 16, 675–679 (2015).
Novakovic, B. et al. β-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 167, 1354–1368.e1314 (2016).
Ostuni, R. et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013).
Schetters, S. T. T., Gomez-Nicola, D., Garcia-Vallejo, J. J. & Van Kooyk, Y. Neuroinflammation: microglia and T cells get ready to tango. Front. Immunol. 8, 1905 (2017).
Lan, X., Han, X., Liu, X. & Wang, J. Inflammatory responses after intracerebral hemorrhage: From cellular function to therapeutic targets. J. Cereb. Blood Flow. Metab. Metab. 39, 184–186 (2019).
Dong, Y. & Yong, V. W. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat. Rev. Neurol. 15, 704–717 (2019).
Chang, C. F., Massey, J., Osherov, A., Angenendt da Costa, L. H. & Sansing, L. H. Bexarotene enhances macrophage erythrophagocytosis and hematoma clearance in experimental intracerebral hemorrhage. Stroke 51, 612–618 (2020).
Zhao, X. et al. Neutrophil polarization by IL-27 as a therapeutic target for intracerebral hemorrhage. Nat. Commun. 8, 602 (2017).
Li, Q. et al. Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: coexistence of ferroptosis, autophagy, and necrosis. Front. Neurol. 9, 581 (2018).
Chen, J. et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 32, 1005–1011 (2001).
Bieber, M. et al. Validity and reliability of neurological scores in mice exposed to middle cerebral artery occlusion. Stroke 50, 2875–2882 (2019).
Leong, A. T. L. et al. Optogenetic auditory fMRI reveals the effects of visual cortical inputs on auditory midbrain response. Sci. Rep. 8, 8736 (2018).
Linfante, I., Llinas, R. H., Caplan, L. R. & Warach, S. MRI features of intracerebral hemorrhage within 2 h from symptom onset. Stroke 30, 2263–2267 (1999).
Lian, H., Roy, E. & Zheng, H. Protocol for primary microglial culture preparation. Bio Protoc. https://doi.org/10.21769/BioProtoc.1989 (2016).
Wang, Y. C. et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 75, 876–889 (2014).
Author information
Authors and Affiliations
Contributions
S.-S.L.: Conceptualized, designed, performed the research, and drafted the manuscript. L.P.: Designed, supervised the research, and critically revised the manuscript. Y.C.: Designed, supervised the research, and reviewed the manuscript. J.X.L. and A.C.K.N. reviewed the manuscript. G.K.K.L.: Conceptualized, designed, supervised the research, and critically revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animal experiments were approved by the Committee on the Use of Live Animals in Teaching and Research of our institution.
Peer review
Peer review information
Communications Biology thanks Shu Wan, Yaohui Tang and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Eve Rogers. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, SS., Pang, L., Cheng, Y. et al. A previous hemorrhagic stroke protects against a subsequent stroke via microglia alternative polarization. Commun Biol 5, 654 (2022). https://doi.org/10.1038/s42003-022-03621-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03621-4
- Springer Nature Limited
This article is cited by
-
Neuroinflammation in Acute Ischemic and Hemorrhagic Stroke
Current Neurology and Neuroscience Reports (2023)