
Abstract
To explore the molecular pathogenesis of pulmonary arterial hypertension (PAH) and identify potential therapeutic targets, we performed transcriptome sequencing of lung tissue from mice with hypoxia-induced pulmonary hypertension. Our Gene Ontology analysis revealed that “extracellular matrix organization” ranked high in the biological process category, and matrix metallopeptidases (MMPs) and other proteases also played important roles in it. Moreover, compared with those in the normoxia group, we confirmed that MMPs expression was upregulated in the hypoxia group, while the hub gene Timp1 was downregulated. Crocin, a natural MMP inhibitor, was found to reduce inflammation, decrease MMPs levels, increase Timp1 expression levels, and attenuate hypoxia-induced pulmonary hypertension in mice. In addition, analysis of the cell distribution of MMPs and Timp1 in the human lung cell atlas using single-cell RNAseq datasets revealed that MMPs and Timp1 are mainly expressed in a population of fibroblasts. Moreover, in vitro experiments revealed that crocin significantly inhibited myofibroblast proliferation, migration, and extracellular matrix deposition. Furthermore, we demonstrated that crocin inhibited TGF-β1-induced fibroblast activation and regulated the pulmonary arterial fibroblast MMP2/TIMP1 balance by inhibiting the TGF-β1/Smad3 signaling pathway. In summary, our results indicate that crocin attenuates hypoxia-induced pulmonary hypertension in mice by inhibiting TGF-β1-induced myofibroblast activation.
Similar content being viewed by others

Introduction
Pulmonary hypertension is a vascular disease associated with severe morbidity and mortality1 that is characterized by pulmonary vascular remodeling and extracellular matrix deposition2. Currently, targeted drugs aimed at relieving pulmonary vasoconstriction have widely used in pulmonary hypertension patients. However, these drugs fail to alleviate pulmonary vascular remodeling and restore right ventricular function, and lung transplantation is ultimately the only curative option3,4. Therefore, identifying the key cells and pathways in pulmonary vascular remodeling may lead to the discovery of new therapeutic targets for pulmonary hypertension.
Recently, studies have used transcriptome sequencing and bioinformatics analysis to screen differentially expressed genes (DEGs) in pulmonary hypertension and further investigate potential biomarkers and regulatory targets. Several transcriptome studies have analyzed the DEGs in cells from patients with idiopathic PH and in the lung tissue of rat models of pulmonary hypertension induced by monocrotaline; these studies have found that DEGs are mainly enriched in the abnormal proliferation of smooth muscle cells and endothelial cells, as well as in inflammatory reactions5,6,11. In addition, Park et al. performed transcriptomic profiling of pulmonary endothelial cells from Sox17-deficient mice and revealed that loss of Sox17 promoted abnormal proliferation and inflammation in lung endothelial cells under hypoxic stress9. Rodor et al. performed scRNA-seq sequencing of lung endothelial cells isolated from an endothelial lineage tracing mouse model and found that 51% of the DEGs were upregulated in rats or human PAH. Although the above transcriptomic studies and sc-RNA-seq data provide insights into PAH development, there are discrepancies in the identified DEGs and results, potentially due to differences in study design, modeling methods, species, intrasample heterogeneity, and data processing software and algorithms. In the present study, we performed a transcriptome study of C57BL/6 mouse HPH to explore the pathogenesis of PAH and identify key molecules and pathways involved. We applied transcriptome sequencing and GO analysis and found that ECM organization, smooth muscle cell proliferation, oxidative stress, leukocyte migration, and inflammatory response play important roles in PAH development.
The composition of the ECM is regulated by the balance between proteolytic enzymes, such as MMPs, metalloproteinases, serine elastase, lysyl oxidase, and their endogenous inhibitors, TIMPs. In PAH, the imbalance of proteolytic enzymes and their endogenous tissue inhibitors leads to increased collagen deposition, collagen crosslinking, and elastin breakdown in the vascular and perivascular compartments of the pulmonary arteries33,34. Benisty et al. reported that the expression of MMPs is significantly increased in the urine of patients with pulmonary hypertension, which may reflect the remodeling of pulmonary vessels35. Soban Umar et al. demonstrated that the activation of MMP signaling in a rat model of pulmonary hypertension promoted ventricular hypertrophy and remodeling36. In the present study, we constructed a PPI network and screened 10 key genes enriched in DEGs from the lung tissues of HPH mice. Among these genes, TIMP1, a component of the endogenous inhibitor metalloproteinase tissue inhibitor, was downregulated. In addition, the expression of MMP-2 and MMP-9 was increased significantly in the mice with HPH, which led to an imbalance in MMPs/TIMP1. Previous studies have shown that this imbalance between MMPs and TIMPs has been proven to induce ECM remodeling in patients with IPAH37. In addition, overexpressing adenovirus TIMP1 in MCT-induced pulmonary hypertension in rats reduced pulmonary vascular remodeling, suggesting that balancing MMPs/TIMP1 can reverse the disease38. However, another study by the same group in hypoxia-induced pulmonary hypertension in rats found that overexpressing adenovirus TIMP1 aggravated pulmonary hypertension. These contradictory results regarding the TIMP1 under hypoxia and monocrotaline pulmonary hypertension model in rats indicated that the beneficial effect of artificially increasing TIMP1 depended on the primary injury involved and its balance with MMPs. In order to determine whether correcting the MMPs/TIMP1 imbalance can ameliorate pulmonary vascular remodeling, we screened potential natural MMP inhibitors. Previous studies have found that two candidate natural compounds, hesperetin and crocin, can inhibit MMP activity29. Among the two candidates, crocin has been reported to attenuate pulmonary inflammation and oxidative stress in a rat model of monocrotaline-induced pulmonary arterial hypertension24,25,26. We confirmed that crocin inhibits pulmonary vascular remodeling and inflammation in HPH, while hesperetin has no protective effect on hypoxia-induced pulmonary hypertension in mice. Regarding the inhibition of hesperetin on pulmonary fibrosis, Li et al. found that hesperetin (200 mg/kg or 400 mg/kg) had a protective effect on silica-induced pulmonary fibrosis; their dose was different from that (50 mg/kg) used in our study. We hypothesize that hesperetin may have protective effects on pulmonary hypertension at a high dose, though further studies with high doses of hesperetin may be needed to confirm this. Additionally, we investigated whether hesperetin affected MMP2/TIMP1 balance in vitro and found that it had no effect on MMP-2/TIMP1 balance (Supplementary Figure 2), which indicates that the mechanism of hesperetin may differ from that of crocin. Previous studies have shown that high doses of crocin proportionally reduce the levels of macrophages and their inflammatory derivatives in atherosclerosis, including MCP-1, TNF-α, IL-6, MMP-2, MMP-3, and MMP-9. In addition, it was found that there was a significant decrease in the MMP-2/TIMP2 ratio after crocin treatment28. Soong et al. reported that crocin inhibited fibroblast proliferation; simultaneously decreased α-SMA expression and the mRNA levels of COL1A1, COL3A1, and MMP-1, and increased Timp1 mRNA levels in bleomycin-induced sclerotic mice, demonstrating the antifibrotic effects of crocin39. Combining these findings with those obtained in our study, we hypothesized that crocin can modulate the balance of MMPs/TIMP1 in the pulmonary tissue of HPH mice.
The role of endothelial cells and smooth muscle cells (SMCs) in vascular remodeling has been extensively studied, but relatively little attention has been given to adventitial fibroblasts40. Fibroblasts are the main producers of the ECM in all organs and play key roles in the coordination of normal tissue homeostasis and the response to disease41. PAF proliferation and differentiation are critical in PAH pathogenesis. Several factors participate in PAF activation. Several studies have shown that the plasma Galectin-3 (Gal-3) level, which is a key fibroblast activation factor, is significantly increased in PAH patients and that Gal-3 expression is upregulated in the adventitia of pulmonary arteries. In addition, inhibition of Gal-3 improved pulmonary vascular remodeling in PAH and simultaneously inhibited the proliferation and differentiation of PAFs42. It was also previously shown that inhibiting FABP5 expression in mice abrogates pulmonary artery remodeling and improves heart function in left heart disease-associated pulmonary hypertension, and silencing FABP5 attenuates the TGF-β1-induced fibrosis response in cultured PAFs43. Chen et al. reported that 5-HT directly activates PAFs and signals through the TGF-β1/Smad 3 pathway to promote fibroblast activation and adventitial fibrosis, ultimately leading to pulmonary hypertension41. Given the above research results, PAF activation likely plays an important role in pulmonary vascular remodeling, and targeting this process may be a new therapeutic approach for treating pulmonary hypertension. Crocin inhibits fibroblast activation and participates in fibrosis in several organs, such as the liver44, lung24,45, and heart46. In the present study, we established a PAF activation model induced by PDGF-BB and clarified whether crocin has an effect on PAF activation. Through BrdU cell proliferation and wound healing cell migration assays, we found that crocin inhibited cell proliferation and migration after administration. Soong et al. reported that crocin inhibited fibroblast proliferation, decreased α-smooth muscle actin (α-SMA) expression, reduced the mRNA levels of COL1A1, COL3A1, and MMP-1, and increased the mRNA levels of Timp1 in bleomycin-induced sclerotic mice, demonstrating the antifibrotic effects of crocin39. To further explore whether crocin affects fibroblast activation, we evaluated the expression levels of α-SMA, Col1a1, COL3A1, and COL5A1 in PAFs. WB revealed that crocin significantly reduced the expression of α-SMA and Col1a1, demonstrating that crocin could inhibit PAF activation.
Dysregulation of TGF-β1 signaling contributes to pulmonary artery remodeling and is thought to promote PAH33, particularly by promoting cell proliferation. Dominant-negative mutation of the TGF-β receptor blocks hypoxia-induced pulmonary artery remodeling in mice47. Activation of TGF-β1 signaling leads to excessive fibroblast proliferation and infiltration, myofibroblast production, extracellular matrix accumulation, and inhibition of collagen degradation33,34. Activation of TGF-β1 induces the phosphorylation of Smad2/3, which forms the Smad complex and interacts with transcription factors, such as α-SMA, to promote gene expression48,49,50. The TGF-β1/SMAD signaling pathway is closely related to cardiovascular diseases51. The TGF-β1/SMAD signaling pathway is one of the major inducers of RV fibrosis in MCT-induced pulmonary hypertension52,53. The above studies suggest that TGF-β1/SMAD3 signaling is involved in PAH development. In the present study, we demonstrated that crocin inhibits fibroblast activation and extracellular matrix production by inhibiting the activation of the TGF-β signaling pathway. WB analysis of lung tissue revealed that activation of the T GF-β1/SMAD3 signaling pathway was significantly inhibited in the crocin group, and activation of the TGF-β1/SMAD3 signaling pathway was significantly inhibited after the administration of crocin in our fibroblast model. To further verify whether fibroblast activation was associated with crocin, we pretreated cells with an agonist of TGF-β1 and then administered crocin. Compared with that in cells not treated with crocin, the inhibitory effect of crocin was partially restored when the agonist was used. Taken together, these findings indicate that crocin modulates TGF-β1/SMAD3 signaling in PAFs, which is the molecular mechanism through which crocin regulates MMP2/TIMP1 homeostasis to inhibit pulmonary vascular remodeling.
Our study has some limitations. We demonstrated the potential protective effect of crocin; however, we did not determine whether crocin can reverse established HPH. In addition, we only applied one dose of crocin in the present study. According to the previously published literature, further studies are needed to determine the optimal dose and ideal therapeutic course of crocin.
Given our results, we conclude that crocin can prevent HPH development in hypoxic mice. We presented new data showing that crocin attenuates pulmonary hypertension, pulmonary vascular remodeling, and RV hypertrophy in HPH mice, likely through blockade of hypoxia-induced hyperactivity of TGF-β1/Smad3 signaling and inhibition of fibroblast activation. This provides a potential therapeutic method for the treatment of pulmonary hypertension in people with chronic hypoxia-related diseases (such as obstructive pulmonary disease, bronchiectasis, altitude sickness, and sleep-related respiratory disorders).
Materials and methods
Hypoxia-induced pulmonary hypertension in mice
Male 6–8-week-old C57BL/6 mice were purchased from HFK Bioscience Company (Bei**g, China), and hypoxia-induced pulmonary hypertension was induced as described previously54. Hesperetin and crocin (Biopurify Phytochemical, Chengdu, China) were intraperitoneally injected at a dose of 50 mg/kg body weight every 3 days. After 4 weeks, the right ventricular systolic pressure (RVSP) was measured, and the right ventricular hypertrophy index was calculated [RVHI = RV/(LV + S) × 100%]. The study was conducted in accordance with the ARRIVE guidelines. The animal experiments in this study were approved by the Bei**g Anzhen Hospital Ethics Committee, and the experimental procedures were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Cell culture
Mouse pulmonary arterial fibroblasts (Procell, Wuhan, China) were maintained in fibroblast medium (ScienCell, San Diego, CA, USA), and SL4 mouse colon cancer cells were maintained in Dulbecco’s modified Eagle medium (DMEM)/F12 (Gibco, New York, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, New York, USA) and 1% penicillin and streptomycin (Gibco, NY, USA) as described previously55. All cells were maintained at 37 °C in 95% humidified air and 5% CO2. Recombinant TGF-β1 (Peprotech, NJ, USA) was used at a dose of 5 ng/ml, recombinant PDGF-BB (MCE, NJ, USA) was used at a dose of 20 ng/ml, and SRI-011381 (MCE, NJ, USA) was used at a dose of 10 μM to treat PAFs.
BrdU assay
Fibroblasts were pretreated with 10 or 50 μM crocin, and then stimulated with PDGF-BB. Then, BrdU (10 μM) was added within 2–4 h before the end of treatment. After 24 h of drug administration, the cells were fixed and incubated with primary BrdU antibody (Zhongshan Golden Bridge, Bei**g, China; 1:200) at 4 °C overnight. Afterward, the cells were incubated with a FITC-labeled secondary antibody (Invitrogen, CA, USA; 1:1000) for 1 h at room temperature. The nuclei were stained with DAPI and detected and analyzed with an ImageXpress XK Microscale (Molecular Devices, CA, USA).
Cell scratch migration assay
Fibroblasts were seeded and scratched with a 200 μL sterile pipette tip 24 h later. The cells were treated with 10 or 50 μM crocin for 30 min, and then stimulated with 20 ng/ml PDGF-BB. The wound area was observed with an inverted light microscope (Leica, Wetzlar, Germany) at 0, 24, and 48 h and was analyzed using ImageJ software (National Institute of Health, MD, USA).
Western blotting (WB)
Lung tissue and cell samples were prepared, and WB was performed as previously described56. The primary antibodies used were as follows: MMP9 (1:1000; Biorbyt, Britain), MMP2 (1:1000; CST, USA), TIMP1 (1:1000; Abcam, USA), MMP3 (1:1000; CST, MA, USA), TGF-β1 (1:1000; Abcam, USA), P-smad3 (1:1000; CST, MA, USA), T-smad3 (1:1000; CST, MA, USA), P-smad2 (1:1000; CST, MA, USA), and T-smad2 (1:1000; CST, MA, USA).
Real-time PCR
Total RNA was extracted from lung tissue and cells using FreZOL reagent (Vazyme, Nan**g, China). cDNA generation and q-PCR analyses were performed using SYBR Greener qPCR SuperMix Universal (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The relative quantification of gene expression (for MMP2, MMP3, MMP9, TIMP1, SPP1, Col1a1, Col3a1, Col5a1, and A-SMA) was determined by comparison with the relative endogenous reference gene GAPDH. The specific primer set sequences are listed in Table 2.
Gelatin zymography assay
MMP activity was measured using a Gelatinase/Collagenase assay kit (Real-Times, Bei**g, China). The cell supernatant was treated and electrophoresed on an 8% sodium dodecyl sulfate‒polyacrylamide gel with 0.1% gelatin. Then, the gel was treated and stained with Coomassie blue R-250 solution as previously described29.
Histopathology
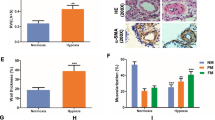
The lung tissues were fixed, embedded, and sectioned as previously described56 and stained with an HE staining kit (Zhongshan Gold Bridge, China). The distal pulmonary artery (with a diameter of 50–150 μm) wall thickness ratio (distal pulmonary artery wall thickness ratio, MWT%) was calculated as follows: MWT% = [(outer pipe diameter)/outer pipe diameter] 100%. The sectioned lung tissues were subjected to Masson staining (Solebo, Bei**g, China) or immunohistochemistry (IHC) with antibodies against TIMP1 (Abcam, Cambridge, UK) and MAC-3 (Santa Cruz Biotechnology, Dallas, TX, USA) as previously described56. For immunofluorescence staining, mouse lung tissues were incubated with antibodies against MMP2, TIMP1, and FSP (Abcam, Cambridge, UK) at 4 °C overnight. Subsequently, FITC- or TRITC-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) were applied at room temperature for 1 h. Images were obtained with a confocal fluorescence microscope (Leica Microsystems, Buffalo Grove, IL, USA).
Transcriptome analysis
Lung tissue RNA was extracted from the hypoxic group (n = 4) and normoxic group (n = 3) using TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Whole-transcriptome sequencing was completed by BGI Genomics Co., Ltd. (Wuhan, China) using the BGISEQ-500 platform. The data were filtered with the filtering software SOAPnuke (version 1.5.2) developed by BGI Genomics.
Protein‒protein interaction (PPI) network analysis and identification of hub genes
The STRING protein database (https://www.string-db.org/) was used for online analysis of the PPI network. Protein interaction data were analyzed with Cytoscape (version 3.8.0) software, the CytoHubba plug-in was used to construct the PPI subnetwork, and the top 10 genes were screened as hub genes according to the topological analysis method of connectivity (degree).
Statistical analysis
Continuous data are presented as the mean ± standard deviation, and a t test was used for the comparison of two independent groups. P < 0.05 was considered to indicate significance. GraphPad Prism 7.0 was used for statistical analyses of the data. The R language and corresponding R software packages were used for bioinformatic analysis and visualization.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) at the National Genomics Data Center (Nucleic Acids Res 2022) and the China National Center for Bioinformation/Bei**g Institute of Genomics, Chinese Academy of Sciences (GSA: CRA015863). They are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
Abbreviations
- CRO:
-
Crocin
- HPH:
-
Hypoxic pulmonary hypertension
- TIMP1:
-
Tissue inhibitor of metalloproteinase
- MMPs:
-
Matrix metallopeptidases
- PDGF-BB:
-
Platelet-derived growth factor-BB
- TGF-β1:
-
Transforming growth factor-β1
- SMAD:
-
Small mothers against decapentaplegic
References
Tuder, R. M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 367, 643–649 (2017).
Umar, S. et al. The Y chromosome plays a protective role in experimental hypoxic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 197, 952–955 (2018).
Tuder, R. M. et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 62, D4-12 (2013).
Thenappan, T., Ormiston, M. L., Ryan, J. J. & Archer, S. L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ (Clin. Res. Ed.) 360, 5492 (2018).
Chen, Y. et al. Identification of immune-related hub genes and analysis of infiltrated immune cells of idiopathic pulmonary artery hypertension. Front. Cardiovasc. Med. 10, 1125063 (2023).
Gorr, M. W., Sriram, K., Muthusamy, A. & Insel, P. A. Transcriptomic analysis of pulmonary artery smooth muscle cells identifies new potential therapeutic targets for idiopathic pulmonary arterial hypertension. Br. J. Pharmacol. 177, 3505–3518 (2020).
**ao, G. et al. RNA sequencing analysis of monocrotaline-induced PAH reveals dysregulated chemokine and neuroactive ligand receptor pathways. Aging (Albany, NY) 12, 4953–4969 (2020).
Xu, S. L. et al. Regulation of circular RNAs act as ceRNA in a hypoxic pulmonary hypertension rat model. Genomics 113, 11–19 (2021).
Park, C. S. et al. Sox17 deficiency promotes pulmonary arterial hypertension via HGF/c-Met signaling. Circ. Res. 131, 792–806 (2022).
de la Cuesta, F. et al. Extracellular vesicle cross-talk between pulmonary artery smooth muscle cells and endothelium during excessive TGF-beta signalling: Implications for PAH vascular remodelling. Cell Commun. Signal 17, 143 (2019).
Ikeda, K. T. et al. Hypoxia-induced pulmonary hypertension in different mouse strains: Relation to transcriptome. Am. J. Respir. Cell Mol. Biol. 60, 106–116 (2019).
Zhang, L. et al. TGF-β1/FGF-2 signaling mediates the 15-HETE-induced differentiation of adventitial fibroblasts into myofibroblasts. Lipids Health Dis. 15, 2 (2016).
Wang, D. et al. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ. Res. 114, 67–78 (2014).
Yang, B. et al. Adventitial transduction of lentivirus-shRNA-VEGF-A in arteriovenous fistula reduces venous stenosis formation. Kidney Int. 85, 289–306 (2014).
Kumar, V., Goutam, R. S., Park, S., Lee, U. & Kim, J. Functional roles of FGF signaling in early development of vertebrate embryos. Cells 10, 2148 (2021).
Srivastava, R., Ahmed, H., Dixit, R.K., Dharamveer & Saraf, S.A. Crocus sativus L.: A comprehensive review. Pharmacognosy Rev. 4, 200–208 (2010).
Pham, T. Q., Cormier, F., Farnworth, E., Tong, V. H. & Van Calsteren, M. R. Antioxidant properties of crocin from Gardenia jasminoides Ellis and study of the reactions of crocin with linoleic acid and crocin with oxygen. J. Agric. Food Chem. 48, 1455–1461 (2000).
Bukhari, S.I., Manzoor, M. & Dhar, M.K. A comprehensive review of the pharmacological potential of Crocus sativus and its bioactive apocarotenoids. Biomedicine & pharmacotherapy Biomedecine pharmacotherapie 98, 733–745 (2018).
Rahaiee, S., Moini, S., Hashemi, M. & Shojaosadati, S. A. Evaluation of antioxidant activities of bioactive compounds and various extracts obtained from saffron (Crocus sativus L.): A review. J. Food Sci. Technol. 52, 1881–1888 (2015).
Soeda, S., Aritake, K., Urade, Y., Sato, H. & Shoyama, Y. Neuroprotective activities of Saffron and Crocin. Adv. Neurobiol. 12, 275–292 (2016).
Boskabady, M. H., Shafei, M. N., Shakiba, A. & Sefidi, H. S. Effect of aqueous-ethanol extract from Crocus sativus (saffron) on guinea-pig isolated heart. Phytother. Res. PTR 22, 330–334 (2008).
Chen, X., Huang, J., Lv, Y., Chen, Y. & Rao, J. Crocin exhibits an antihypertensive effect in a rat model of gestational hypertension and activates the Nrf-2/HO-1 signaling pathway. Hypertens. Res. 44, 642–650 (2021).
Imenshahidi, M., Hosseinzadeh, H. & Javadpour, Y. Hypotensive effect of aqueous saffron extract (Crocus sativus L.) and its constituents, safranal and crocin, in normotensive and hypertensive rats. Phytother. Res. 24, 990–994 (2010).
Zaghloul, M. S., Said, E., Suddek, G. M. & Salem, H. A. Crocin attenuates lung inflammation and pulmonary vascular dysfunction in a rat model of bleomycin-induced pulmonary fibrosis. Life Sci. 235, 116794 (2019).
Dianat, M., Radan, M., Mard, S. A., Sohrabi, F. & Saryazdi, S. S. N. Contribution of reactive oxygen species via the OXR1 signaling pathway in the pathogenesis of monocrotaline-induced pulmonary arterial hypertension: The protective role of Crocin. Life Sci. 256, 117848 (2020).
Sheng, Y., Gong, X., Zhao, J., Liu, Y. & Yuan, Y. Effects of crocin on CCL2/CCR2 inflammatory pathway in monocrotaline-induced pulmonary arterial hypertension rats. Am. J. Chin. Med. 50, 241–259 (2022).
Lu, P. et al. Antitumor effects of crocin on human breast cancer cells. Int. J. Clin. Exp. Med. 8, 20316–20322 (2015).
Christodoulou, E. et al. Crocus sativus L. aqueous extract reduces atherogenesis, increases atherosclerotic plaque stability and improves glucose control in diabetic atherosclerotic animals. Atherosclerosis 268, 207–214 (2018).
Qi, F. et al. Artificial intelligence uncovers natural MMP inhibitor crocin as a potential treatment of thoracic aortic aneurysm and dissection. Front Cardiovasc. Med. 9, 871486 (2022).
Hu, C. J., Zhang, H., Laux, A., Pullamsetti, S. S. & Stenmark, K. R. Mechanisms contributing to persistently activated cell phenotypes in pulmonary hypertension. J. Physiol. 597, 1103–1119 (2019).
Spaczynska, M., Rocha, S. F. & Oliver, E. Pharmacology of pulmonary arterial hypertension: An overview of current and emerging therapies. ACS Pharmacol. Transl. Sci. 3, 598–612 (2020).
Rodor, J. et al. Single-cell RNA sequencing profiling of mouse endothelial cells in response to pulmonary arterial hypertension. Cardiovasc. Res. 118, 2519–2534 (2022).
Ren, L. L. et al. Transforming growth factor-β signaling: From tissue fibrosis to therapeutic opportunities. Chemico-Biol. Interact. 369, 110289 (2023).
Poniatowski, ŁA., Wojdasiewicz, P., Gasik, R. & Szukiewicz, D. Transforming growth factor Beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediators Inflamm. 2015, 137823 (2015).
Benisty, J. I. et al. Matrix metalloproteinases in the urine of patients with pulmonary arterial hypertension. Chest 128, 572S (2005).
Umar, S. et al. Activation of signaling molecules and matrix metalloproteinases in right ventricular myocardium of rats with pulmonary hypertension. Pathol. Res. Pract. 203, 863–872 (2007).
Lepetit, H. et al. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 25, 834–842 (2005).
Vieillard-Baron, A. et al. Inhibition of matrix metalloproteinases by lung TIMP-1 gene transfer limits monocrotaline-induced pulmonary vascular remodeling in rats. Hum. Gene Ther. 14, 861–869 (2003).
Song, Y., Zhu, L. & Li, M. Antifibrotic effects of crocetin in scleroderma fibroblasts and in bleomycin-induced sclerotic mice. Clinics (Sao Paulo, Brazil) 68, 1350–1357 (2013).
Stenmark, K. R., Bouchey, D., Nemenoff, R., Dempsey, E. C. & Das, M. Hypoxia-induced pulmonary vascular remodeling: Contribution of the adventitial fibroblasts. Physiol. Res. 49, 503–517 (2000).
Chen, C. et al. Serotonin drives the activation of pulmonary artery adventitial fibroblasts and TGF-β1/Smad3-mediated fibrotic responses through 5-HT(2A) receptors. Mol. Cell. Biochem. 397, 267–276 (2014).
Luo, H. et al. Galectin-3 mediates pulmonary vascular remodeling in hypoxia-induced pulmonary arterial hypertension. J. Am. Soc. Hypertens. JASH 11, 673-683.e673 (2017).
Lei, Q., Yu, Z., Li, H., Cheng, J. & Wang, Y. Fatty acid-binding protein 5 aggravates pulmonary artery fibrosis in pulmonary hypertension secondary to left heart disease via activating wnt/β-catenin pathway. J. Adv. Res. 40, 197–206 (2022).
Xuan, J. et al. Crocin inhibits the activation of mouse hepatic stellate cells via the lnc-LFAR1/MTF-1/GDNF pathway. Cell Cycle 19, 3480–3490 (2020).
Mehrabani, M. et al. Crocin: A protective natural antioxidant against pulmonary fibrosis induced by bleomycin. Pharmacol. Rep. 72, 992–1001 (2020).
**, W. et al. Crocin attenuates isoprenaline-induced myocardial fibrosis by targeting TLR4/NF-kappaB signaling: Connecting oxidative stress, inflammation, and apoptosis. Naunyn Schmiedebergs Arch. Pharmacol. 393, 13–23 (2020).
Chen, Y. F. et al. Dominant negative mutation of the TGF-beta receptor blocks hypoxia-induced pulmonary vascular remodeling. J. Appl. Physiol. 1985(100), 564–571 (2006).
Khalil, H. et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 127, 3770–3783 (2017).
Piersma, B., Bank, R. A. & Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2, 59 (2015).
Dai, J. et al. Bi-directional regulation of TGF-β/Smad pathway by arsenic: A systemic review and meta-analysis of in vivo and in vitro studies. Life Sci. 220, 92–105 (2019).
Zelarayan, L. C., Noack, C., Zafiriou, M. P., Renger, A. & Bergmann, M. W. Wnt signaling molecules in left ventricular remodeling: Focus on dishevelled 1. Hypertension (Dallas, Tex.:1979) 55, 852–854 (2010).
Sanada, T. J. et al. Altered TGFbeta/SMAD signaling in human and rat models of pulmonary hypertension: An old target needs attention. Cells 10, 1–13 (2021).
Krzyżewska, A., Baranowska-Kuczko, M., Kasacka, I. & Kozłowska, H. Cannabidiol alleviates right ventricular fibrosis by inhibiting the transforming growth factor β pathway in monocrotaline-induced pulmonary hypertension in rats. Biochimica et biophysica acta. Mol. Basis Dis. 1869, 166753 (2023).
**, X. et al. SGK1 mediates hypoxic pulmonary hypertension through promoting macrophage infiltration and activation. Anal Cell Pathol. (Amst.) 2019, 3013765 (2019).
Yang, M. et al. Tumor cell-activated CARD9 signaling contributes to metastasis-associated macrophage polarization. Cell Death Differ. 21, 1290–1302 (2014).
Piao, C. et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell Res. 366, 127–138 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Numbers: 82170409, 81800222).
Author information
Authors and Affiliations
Contributions
C.P., J.D., and R.Q.W. contributed to the conception and design of the study, performed experiments, analyzed the data, and drafted the manuscript; Z.W.M., X.D.W., C.Y.S, Y.R., and J.D. participated in data analysis and provided materials; and M.J. and C.P. provided mentorship and revised the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, J., Wei, RQ., Zhang, WM. et al. Crocin's role in modulating MMP2/TIMP1 and mitigating hypoxia-induced pulmonary hypertension in mice. Sci Rep 14, 12716 (2024). https://doi.org/10.1038/s41598-024-62900-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-62900-8
- Springer Nature Limited