
Abstract
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in developed countries, characterized by the death of retinal pigment epithelial (RPE) cells and photoreceptors. Previous studies report an accumulation of damaged and dysfunctional mitochondria in RPE of human donors with AMD. Understanding how damaged mitochondria accumulate in AMD is an important step in discovering disease mechanisms and identifying therapeutic targets. In this report, we assessed mitochondrial fission and fusion by quantifying proteins and measured mitochondrial autophagy (mitophagy) via protein analysis and advanced imaging techniques using mitochondrial targeted mKeima in primary human RPE from donors with or without AMD. We report disease-specific differences in mitochondrial proteins that regulate fission, fusion, and mitophagy that were present at baseline and with treatments to stimulate these pathways. Data suggest AMD RPE utilize receptor-mediated mitophagy as a compensatory mechanism for deficits in the ubiquitin-mediated mitophagy pathway. These changes in mitochondrial homeostasis could lead to the buildup of damaged and dysfunctional mitochondria observed in the RPE of AMD donors.
Similar content being viewed by others

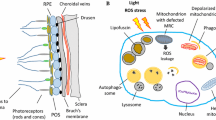
Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in developed countries. This multifactorial disease involves complex genetic and environmental factors whose effects accelerate with age1,2. AMD affects 30% of individuals 75–85 years of age, with a global estimate of 128 million cases in 2020 and 288 million cases by 2040, a consequence of the world’s increasing elderly population3,4. The two forms of AMD include “wet” and “dry”, with wet AMD caused by the abnormal growth of blood vessels into the retina. While wet AMD is less common, it has a number of effective therapies to halt or prevent vision loss5. Dry AMD is the most common form of the disease, accounting for approximately 85% of all AMD cases6. Vision loss associated with dry AMD is caused by the death of retinal pigment epithelium (RPE) and photoreceptors. RPE are essential for maintaining a healthy retina as they are responsible for key functions, such as the transport of nutrients to photoreceptors and the directed secretion of growth factors7. Currently, there are no effective treatments for dry AMD, due to our incomplete understanding of the cellular events causing disease pathology. Thus, there remains an urgent need to identify the underlying mechanisms causing AMD in order to successfully develop therapeutic interventions.
One of the prevailing hypotheses is that RPE mitochondrial defects drive AMD pathology8. This hypothesis is supported by numerous studies in human retina from eye bank donors. Analysis of electron microscopy images found donors with AMD had significantly fewer mitochondrial number, reduced surface area, and an altered cristae morphology9. Additionally, proteomic studies of human RPE tissue found an altered mitochondrial proteome, with multiple proteins in the electron transport chain decreasing in AMD RPE10,11. There are also reports of increased mitochondrial DNA damage with progression of AMD severity12,13. Consistent with the mitochondrial defects observed in tissue, cultures of human primary RPE showed significantly decreased mitochondrial function in RPE from donors with AMD14,46. Additional compounds designed to protect mitochondria from oxidative damage (N-acetyl-L-Cysteine; NAC), remove damaged mitochondria via increased autophagy (rapamycin), upregulate mitochondrial biogenesis (pyrroloquinoline; PQQ), or improve oxidative phosphorylation (nicotinamide mononucleotide, NMN) have been investigated in our lab47. We found that RPE from AMD donors responded to the drugs as detected by the increase in mitochondrial function. In contrast, RPE from donors without AMD did not respond to the drugs. These results are consistent with the idea that mitochondrial dysfunction in the diseased cells can be ameliorated by treatments that target mitochondrial defects. Our study supports the idea that mitochondrial defects drive AMD, and targeting pathways of mitochondrial homeostasis may be a viable treatment option.
This study uncovers potential mechanisms leading to mitochondrial damage and dysfunction associated with AMD that may initiate the metabolic crisis in the retina. We observed disease-specific differences under basal conditions and in response to two mitochondrial stressors, FCCP and CoCl2. The AMD-associated changes in mitochondrial proteins reported in this study could lead to the buildup of damaged and dysfunctional mitochondria that begins to disrupt the delicate retinal ecosystem and lead to the eventual death of RPE and photoreceptors in AMD. Experiments using different stressors and analytical methods may further identify mechanisms that explain these AMD-associated changes in mitochondrial homeostasis, providing new therapeutic targets to treat AMD.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Bowes Rickman, C., Farsiu, S., Toth, C. A. & Klingeborn, M. Dry Age-Related Macular Degeneration: Mechanisms, Therapeutic Targets, and Imaging. Invest. Opthalmol. Visual Sci. 54, ORSF68 (2013).
Kaarniranta, K. et al. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 79, 100858 (2020).
Wong, W. L. et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob. Health 2, e106–e116 (2014).
Leibowitz, H. M. et al. The Framingham Eye Study monograph: An ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv. Ophthalmol. 24, 335–610.
Treatments for Wet AMD (Advanced Neovascular AMD) | National Eye Institute. https://www.nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases/age-related-macular-degeneration/wet-amd-type-late-age-related-macular-degeneration-read-about-treatments-wet-amd-anti-vegf.
Seddon, J. M. & Chen, C. A. The epidemiology of age-related macular degeneration. Int. Ophthalmol. Clin. 44, 17–39 (2004).
Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 85, 845–881 (2005).
Fisher, C. R. & Ferrington, D. A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Invest. Opthalmol. Visual Sci. 59, AMD41 (2018).
Feher, J. et al. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 27, 983–993 (2006).
Nordgaard, C. L., Karunadharma, P. P., Feng, X., Olsen, T. W. & Ferrington, D. A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest. Opthalmol. Visual Sci. 49, 2848 (2008).
Nordgaard, C. L. et al. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest. Opthalmol. Visual Sci. 47, 815 (2006).
Ferrington, D. A. et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 145, 269–277 (2016).
Karunadharma, P. P., Nordgaard, C. L., Olsen, T. W. & Ferrington, D. A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest. Opthalmol. Visual Sci. 51, 5470 (2010).
Ferrington, D. A. et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 13, 255–265 (2017).
Golestaneh, N., Chu, Y., **ao, Y. Y., Stoleru, G. L. & Theos, A. C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 8, (2017).
Cano, M. et al. Nrf2 deficiency decreases NADPH from impaired IDH shuttle and pentose phosphate pathway in retinal pigmented epithelial cells to magnify oxidative stress‐induced mitochondrial dysfunction. Aging Cell 20, (2021).
Fisher, C. R., Ebeling, M. C. & Ferrington, D. A. Quantification of mitophagy using mKeima-mito in cultured human primary retinal pigment epithelial cells. Exp. Eye Res. 217, 108981 (2022).
Zhang, W. et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5, (2016).
Yu, Y. et al. Inhibition of autophagy enhanced cobalt chloride-induced apoptosis in rat alveolar type II epithelial cells. Mol. Med. Rep. https://doi.org/10.3892/mmr.2018.9209 (2018).
Chen, R. et al. Effects of cobalt chloride, a hypoxia-mimetic agent, on autophagy and atrophy in skeletal C2C12 myotubes. Biomed. Res. Int. 2017, 1–9 (2017).
He, Y. et al. CoCl2 induces apoptosis via a ROS-dependent pathway and Drp1-mediated mitochondria fission in periodontal ligament stem cells. Am. J. Physiol. Cell Physiol. 315, C389–C397 (2018).
Wei, H., Liu, L. & Chen, Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochimica et Biophysica Acta (BBA)—Mol. Cell Res. 1853, 2784–2790 (2015).
Decanini, A., Nordgaard, C. L., Feng, X., Ferrington, D. A. & Olsen, T. W. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol. 143, 607-615.e2 (2007).
Olsen, T. W. & Feng, X. The minnesota grading system of eye bank eyes for age-related macular degeneration. Invest. Opthalmol. Visual Sci. 45, 4484 (2004).
Ebeling, M. C. et al. Impaired mitochondrial function in iPSC-retinal pigment epithelium with the complement factor H polymorphism for age-related macular degeneration. Cells 10, 789 (2021).
Tilokani, L., Nagashima, S., Paupe, V. & Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 62, 341–360 (2018).
del Dotto, V., Fogazza, M., Carelli, V., Rugolo, M. & Zanna, C. Eight human OPA1 isoforms, long and short: What are they for?. Biochim Biophys Acta Bioenerg 1859, 263–269 (2018).
del Dotto, V. et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 19, 2557–2571 (2017).
Twig, G. & Shirihai, O. S. The interplay between mitochondrial dynamics and mitophagy. Antioxidants and Redox Signaling 14 (2011).
Yoon, Y. H. et al. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest. Ophthalmol. Visual Sci. 51, (2010).
Mauthe M, Orhon I, Rocchi C et al. Chloroquine inhitis autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14(8), 1435–1455.
Wu, H. & Chen, Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid. Redox Signal. 22, 1032–1046 (2015).
la Cunza, N. et al. Mitochondria-dependent phase separation of disease-relevant proteins drives pathological features of age-related macular degeneration. JCI Insight 6, (2021).
Otera, H. et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158 (2010).
Mitter, S. K. et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 10, (2014).
Parganlija, D. et al. Loss of PINK1 impairs stress-induced autophagy and cell survival. PLoS ONE 9, e95288 (2014).
Lutz, A. K. et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951 (2009).
Dagda, R. K. et al. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855 (2009).
**ao, B. et al. Reactive oxygen species trigger Parkin/PINK1 pathway–dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 292, (2017).
Koentjoro, B., Park, J.-S. & Sue, C. M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 7, 44373 (2017).
Ferrington, D. A., Fisher, C. R. & Kowluru, R. A. Mitochondrial defects drive degenerative retinal diseases. Trends Mol. Med. 26 (2020).
Ethen, C. M., Reilly, C., Feng, X., Olsen, T. W. & Ferrington, D. A. The proteome of central and peripheral retina with progression of age-related macular degeneration. Invest. Opthalmol. Visual Sci. 47, 2280 (2006).
Terluk, M. R. et al. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 35, 7304–7311 (2015).
Kanow, M. A. et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife 6, (2017).
ReCLAIM-2 study to evaluate safety, efficacy & pharmacokinetics of elamipretide in subjects with AMD With Non-central GA - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03891875.
Brown, E. E. et al. The common antidiabetic drug metformin reduces odds of develo** age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 60, 1470–1477 (2019).
Ebeling, M. C. et al. Improving retinal mitochondrial function as a treatment for age-related macular degeneration. Redox. Biol. 34, 101552 (2020).
Acknowledgements
The authors wish to acknowledge the contribution of personnel from the Lions Gift of Sight (St. Paul, MN) for their assistance in procuring human donor eyes and processing eye tissue. The authors also thank the donors and their families for their essential contributions to the research. This work was supported in part by the National Institutes of Health (NIH) National Eye Institute (NEI) F31-EY031558 (to CRF), T32-EY025187 (to CRF), R01EY026012 (to DAF), and R01EY028554 (to DAF), NIH National Institute of Aging (NIA) T32-AG029796 (to CRF), Diana Jacobs Kalman/AFAR Scholarships for Research in the Biology of Aging (to CRF), VitreoRetinal Surgery Foundation Fellowship (to CRF), University of Minnesota Undergraduate Research Opportunities Program (to AAS), the Elaine and Robert Larson Endowed Vision Chair, the Lindsay Family Foundation, and an anonymous benefactor for AMD research.
Author information
Authors and Affiliations
Contributions
Conceptualization: C.R.F., A.A.S., D.A.F.; Methodology: C.R.F., A.A.S., M.C.E., D.A.F.; Investigation: C.R.F., A.A.S., M.C.E., S.R.M.; Writing—Original Draft: C.R.F.; Writing—Review and Editing: C.R.F., A.A.S., M.C.E., S.R.M., D.A.F.; Supervision: C.R.F. and D.A.F.; Funding Acquisition: C.R.F. and D.A.F.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fisher, C.R., Shaaeli, A.A., Ebeling, M.C. et al. Investigating mitochondrial fission, fusion, and autophagy in retinal pigment epithelium from donors with age-related macular degeneration. Sci Rep 12, 21725 (2022). https://doi.org/10.1038/s41598-022-26012-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26012-5
- Springer Nature Limited
This article is cited by
-
Urolithin A promotes p62-dependent lysophagy to prevent acute retinal neurodegeneration
Molecular Neurodegeneration (2024)