
Abstract
The first synthetic cannabinoid receptor agonists (SCRAs) were designed as tool compounds to study the endocannabinoid system’s two predominant cannabinoid receptors, CB1R and CB2R. Unfortunately, novel SCRAs now represent the most rapidly proliferating novel psychoactive substances (NPS) of abuse globally. Unlike ∆9-tetrahydrocannabinol, the CB1R and CB2R partial agonist and the intoxicating constituent of Cannabis, many SCRAs characterized to date are full agonists of CB1R. Gaining additional insight into the pharmacological activity of these SCRAs is critical to assess and regulate NPSs as they enter the marketplace. The purpose of this study was to assess select SCRAs recently identified by Canadian police, border service agency, private companies and the illicit market as potential CB1R and CB2R agonists. To this end, fifteen SCRAs were screened for in vitro activity and in silico interactions at CB1R and CB2R. Several SCRAs were identified as being highly biased for cAMP inhibition or βarrestin2 recruitment and receptor subtype selectivity between CB1R and CB2R. The indazole ring and halogen-substituted butyl or pentyl moieties were identified as two structural features that may direct βarrestin2 bias. Two highly-biased SCRAs—JWH-018 2′-napthyl-N-(3-methylbutyl) isomer (biased toward cAMP inhibition) and 4-fluoro MDMB-BINACA (biased toward βarrestin2 recruitment) displayed unique and differential in vivo activity in mice. These data provide initial insight into the correlations between structure, signalling bias, and in vivo activity of the SCRAs.
Similar content being viewed by others

Introduction
In 2018 Canada became the first G7 country to legalize Cannabis sativa for medical and recreational purposes. It is critically important that we gain a more comprehensive understanding of cannabinoid pharmacology in order to reduce harm and make full use of this plant’s medical potential. Biologically active compounds in Cannabis are referred to as ‘phytocannabinoids’. The two best-known phytocannabinoids are ∆9-tetrahydrocannabinol (THC) and cannabidiol (CBD). Our bodies also naturally produce endogenous cannabinoids anandamide (AEA) and 2-arachidonoylglycerol (2-AG). These cannabinoids act to modulate the body’s cannabinoid receptors: CB1R and CB2R, limiting neurotransmitter release throughout the brain and inflammatory processes, respectively 1. Beyond these naturally occurring compounds, a wide and structurally diverse array of synthetic cannabinoid receptor agonists (SCRAs) have been produced (Fig. 1) 1. These were originally intended to aid in drug development and as tool compounds to help better understand the body’s endogenous cannabinoid system 1,2. Unfortunately, many of these compounds are now available through illegal markets where they are sold as psychoactive and intoxicating drugs of misuse known colloquially as ‘spice’ or ‘K-2’1,2.

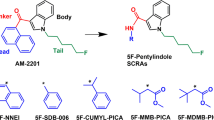
Structures of SCRAs examined in this study. CP55,940 and ∆9-THC were used as reference ligands. Chemical structures were drawn in Microsoft PowerPoint by the authors.
SCRAs pose serious health risks to Canadians as they can lead to severe vomiting, chest pain, increased heart rate, vision blackouts, headaches, kidney damage, agitation, high blood pressure, and psychosis 3,4. Acute toxicities of SCRAs such as JWH 018 and JWH 073 are thought to be mediated by CB1R activation 5,6,7; however, novel SCRAs continue to emerge on the market without previous characterization in the scientific literature 4. In Canada, the Controlled Drugs and Substances Act (CDSA) provides a legislative framework for the control of substances that can alter mental processes and that may cause harm to the health of an individual or society when misused or diverted to the illegal market. Many SCRAs are controlled under Item 2 of Schedule II to the CDSA under the heading “Synthetic cannabinoid receptor-type one agonists, their salts, derivatives, isomers, and salts of derivatives and isomers”.
The cannabinoid receptors are activated by a wide spectrum of structurally-diverse cannabinoids 1. Although classically considered Gαi/o-coupled receptors, CB1R and CB2R are a pleiotropically-coupled GPCRs that modulate intracellular signalling through βarrestins and other G proteins 8,9,10,11,12,13. Gαi/o-dependent signalling inhibits cAMP accumulation 8. In contrast, recruitment of βarrestin1 and βarrestin2 to CB1R results in receptor internalization, recycling or turnover 11,12,13. Ligand bias describes a compound’s ability to promote a certain receptor conformation that favours the preferential coupling with, and signalling through, a specific effector protein relative to other effector proteins 11. CB1R and CB2R ligand bias has been observed in multiple cell culture systems and with many SCRAs 11,12,13. Previously, we demonstrated that βarrestin1-biased cannabinoids, such as CP55940 and THC, reduce cellular viability in a cell culture model of Huntington’s disease 11. Our data, and the data of other groups 14,15,16,17,18,19, indicate that cannabinoid receptor ligand bias has unique cell-, and potentially organism-level, effects that may explain the unique side effect profiles of SCRAs as compared to Cannabis-derived cannabinoids.
SCRAs are one of the most rapidly growing classes of novel psychoactive substances (NPS) and are used as recreational drugs in Canada. Unlike the partial agonist THC, many SCRAs are full agonists of CB1R, which may contribute to these drug’s side effect profile 1,2. Recently, several studies have assessed the signaling bias and kinetics of several SCRAs at CB1R through in vitro assays 14,15,16,17,18,19. These studies have revealed that SCRAs with shared structural features, such as indazole rings, are capable of having high affinity, potency, and efficacy at CB1R. For example, Patel et al. 17 observed that eleven such SCRAs are highly potent and efficacious unbiased CB1R agonists. This existing work provides evidence that differentiates SCRAs from partial agonists such as THC because indazole-containing SCRAs produce a more stabilizing effect on the “twin toggle switch” that affords CB1R-Gαi interactions as compared to less-stable interactions between the twin toggle switch and THC 17,20. It is hoped that the growing body of SCRA literature will afford a better understanding of the structure–activity correlates that underlie toxicities unique to SCRA use as compared to Cannabis. Because novel SCRAs with unknown or poorly understood pharmacology continue to be synthetized by illegal markets, gaining additional insight into the pharmacodynamics of these SCRAs is critical to assess the risks of these novel compounds as they enter the marketplace.
The purpose of this study was to assess select emerging SCRAs for their potency, efficacy, binding affinity and differential signaling via CB1R and CB2R. Fifteen SCRAs were assessed in vitro for binding, inhibition of forskolin (FSK)-stimulated cAMP accumulation, and βarrestin2 recruitment at CB1R and CB2R; and in silico to model their unique interactions with CB1R and CB2R. Based on their unique ligand bias, two SCRAs were further assessed in vivo to determine whether observed signaling bias in vitro correlated with specific in vivo drug responses. Our data add to the growing understanding of SCRA structure–activity relationships (SAR) and may extend this SAR into correlations with ligand bias in vitro and in vivo.
Results
Type 1 cannabinoid receptor (CB1R)
Radioligand binding
Saturation binding, including non-specific binding performed with CHO-K1 cells not expressing hCB1R, was conducted with [3H]CP55,940 to determine Kd (0.79 [0.24–2.7] nM) and Bmax (1344 ± 32 fmol/mg) (Supplementary Fig. 1a). All fifteen SCRAs displaced 1.0 nM [3H]CP55,940 from CB1R. ∆9-THC was assayed as a control ligand and displayed similar affinity to that of CP55940 (Fig. 2, Table 1). Ten SCRAs displayed affinity (Ki) that was not different from that of the reference agonist CP55940 at CB1R (Fig. 2, Table 1). Five compounds—JWH 182, JWH 018 2′-naphthyl-N-(3-methylbutyl) isomer, AM 2232, FDU-NNEI, and Cl 2201 displayed significantly greater affinity than that of CP55,940 for CB1R; and one compound—5-fluoro MPP-PICA displayed significantly less affinity than that of CP55,940 for CB1R (Fig. 2, Table 1). Five compounds were able to fully compete 1.0 nM [3H]CP55,940 from CB1R: JWH 182, JWH 018 2′-naphthyl-N-(3-methylbutyl) isomer, MAM2201 N-(5-chloropentyl) analog, ortho-AB-FUBINACA, and FDU-NNEI (Fig. 2, Table 1); all other SCRAs only partially displaced [3H]CP55,940 (22–77%).

Effect of SCRAs on [3H]CP55,940 binding to CB1R. 1 nM [3H]CP55,940 to membranes obtained from CHO cells stably-expressing hCB1R. Data were fit to a nonlinear regression (four parameter model, GraphPad v. 8) for statistics in Table 1. Data are mean ± S.E.M. n ≥ 6 independent experiments performed in duplicate.
Inhibition of FSK-stimulated cAMP
All fifteen SCRAs displayed agonist activity for the inhibition of FSK-stimulated cAMP at CB1R. The majority of these were full agonists of CB1R-dependent cAMP inhibition relative to the reference agonist CP55940, and displayed similar potency compared to that of CP55940 as well (Fig. 3, Table 1). ∆9-THC was assayed as a control partial agonist and displayed significantly lower efficacy and potency relative to CP55940 (Fig. 3, Table 1). One SCRA—AB-PINACA N-(2-fluoropentyl) isomer—was a partial agonist of CB1R-dependent cAMP inhibition relative to CP55940, but retained high potency (Fig. 3, Table 1). Two compounds—AM 2232 and EG 018—displayed significantly lower potency relative to CP55940, but retained full (i.e., 100%) efficacy (Fig. 3, Table 1). Importantly, cAMP accumulation was not completely inhibited by the maximum effects observed with SCRAs, indicating that supramaximal responses could have been detected had they occurred (Supplementary Fig. 2a).

CB1R-dependent inhibition of FSK-stimulated cAMP following SCRA treatment. CHO cells stably-expressing hCB1R were treated with 10 µM FSK and 0.10 nM–10 μM SCRA for 90 min and inhibition of cAMP was measured. cAMP inhibition data are expressed as % CP55,940 response. Data were fit to a nonlinear regression (four parameter model, GraphPad v. 8) for statistics in Table 1; or fit to the operational model to calculate bias (Fig. 5). Data are mean ± S.E.M. n ≥ 6 independent experiments performed in triplicate.
βarrestin2 recruitment
Nine of fifteen SCRAs displayed remarkably weak (i.e. low efficacy and potency > 10,000 nM) agonist activity for the recruitment of βarrestin2 to CB1R relative to CP55940 (Fig. 4, Table 1). ∆9-THC was assayed as a control partial agonist and displayed significantly lower efficacy, but similar potency, relative to CP55940 (Fig. 4, Table 1). JWH 182, ortho-AB-FUBINACA, and 5-fluoro NPB-22 were equipotent, but produced supramaximal EMax responses (i.e. > 100%) relative to CP55940 (Fig. 4, Table 1). AB-PINACA N-(2-fluoropentyl) isomer, 3-chloro AB-PINACA, and 4-fluoro MPP-PICA displayed greater potency and produced supramaximal EMax responses (i.e. > 100%) relative to CP55940 (Fig. 4, Table 1).

βarrestin2 recruitment to CB1R following SCRA treatment. CHO cells stably-expressing hCB1R were treated with 0.10 nM–10 μM SCRA 90 min and βarrestin2 recruitment was measured. βarrestin2 recruitment data are expressed as % CP55,940 response. Data were fit to a nonlinear regression (four parameter model, GraphPad v. 8) for statistics in Table 1; or fit to the operational model to calculate bias (Fig. 5). Data are mean ± S.E.M. n ≥ 6 independent experiments performed in triplicate. Note the difference in y-axis scale between (a) and all other panels.
Bias analyses
Given the high potency and efficacy observed for most SCRAs in the cAMP inhibition assay and comparatively low potency and efficacy observed for the majority of these compounds in the βarrestin2 recruitment assay. It is not surprising that most of the SCRAs tested displayed bias toward the inhibition of cAMP relative to βarrestin2 recruitment (i.e. ∆∆LogR > 0) (Fig. 5). Two SCRAs—AB-PINACA N-(2-fluoropentyl) isomer, 3-chloro AB-PINACA did not display a bias for cAMP inhibition versus βarrestin2 recruitment (i.e. ∆∆LogR not different from 0) (Fig. 5). Finally, three compounds—ortho-AB-FUBINACA, 4-fluoro MDMB-BINACA, and 5-fluoro NPB-22 displayed a significant bias toward the recruitment of βarrestin2 relative to inhibition of FSK-stimulated cAMP (i.e. ∆∆LogR < 0) (Fig. 5).

CB1R SCRA bias. Compound bias between cAMP inhibition and βarrestin2 recruitment [ΔΔLogR (cAMP—βarr2)] is shown here for data presented in Figs. 3 and 4; Table 1. Data were fit to the operational model to calculate bias. Data are mean with 95% CI, n ≥ 6 independent experiments performed in triplicate. Values > 0 represent cAMP bias (white), values < 0 represent βarrestin2 bias (red), and values not different from 0 are unbiased (blue) as determined by 95% CI not overlap** with 0.
Type 2 cannabinoid receptor (CB2R)
Radioligand binding
Saturation binding in CHO-K1 cells stably expressing hCB2R, as well as non-specific binding performed with CHO-K1 cells not expressing hCB2R, was conducted with [3H]CP55,940 to determine Kd (0.93 [0.29–1.9] nM) and Bmax (1447 ± 50 fmol/mg) (Supplementary Fig. 1b). All fifteen SCRAs displaced 1.0 nM [3H]CP55,940 from CB2R. ∆9-THC was assayed as a control ligand and did fully compete [3H]CP55,940 from the receptor (Fig. 6, Table 1). Six SCRAs displayed affinity (Ki) that was not different from that of the reference agonist CP55,940 at CB2R (Fig. 6, Table 1). Four compounds (meta-AB-FUBINACA, ortho-AB-FUBINACA, EG 018, and 4-fluoro MDMB-BINACA) displayed significantly greater affinity than that of CP55,940 for CB2R; and five compounds (MAM2201 N-(5-chloropentyl) analog, AM 2232, AB-PINACA N-(2-fluoropentyl) isomer, JWH 145, and 5-fluoro MPP-PICA) displayed significantly less affinity than that of CP55,940 for CB2R (Fig. 6, Table 1). Of these, MAM2201 N-(5-chloropentyl) analog displayed very low affinity (> 10,000 nM). Only two SCRAs completely displaced 1.0 nM [3H]CP55,940 from CB2R: ortho-AB-FUBINACA and AB-PINACA N-(2-fluoropentyl) isomer (Fig. 6, Table 1); all other SCRAs only partially displaced [3H]CP55,940 (21–89%).

Effect of SCRAs on [3H]CP55,940 binding to CB2R. 1 nM [3H]CP55,940 to membranes obtained from CHO cells stably-expressing hCB2R. Data were fit to a nonlinear regression (four parameter model, GraphPad v. 8) for statistics in Table 1. Data are mean ± S.E.M. n ≥ 6 independent experiments performed in duplicate.
Inhibition of FSK-stimulated cAMP
All fifteen SCRAs displayed full agonist activity to inhibit FSK-stimulated cAMP at CB2R (i.e. not different from 100%) (Fig. 7, Table 1). With one exception, MAM2201 N-(5-chloropentyl) analog, the potency of these compounds at CB2R for cAMP inhibition was not different compared to that of CP55,940 (Fig. 7, Table 1). ∆9-THC was assayed as a control partial agonist and displayed significantly lower efficacy relative to CP55,940 (Fig. 7, Table 1). As was observed for CB1R, cAMP accumulation was not completely inhibited by the maximum effects observed with SCRAs, indicating that supramaximal responses could have been detected had they occurred (Supplementary Fig. 2b).

CB2R-dependent inhibition of FSK-stimulated cAMP following SCRA treatment. CHO cells stably-expressing hCB2R were treated with 10 µM FSK and 0.10 nM–10 μM SCRA for 90 min, and inhibition of cAMP was measured. cAMP inhibition data are expressed as % CP55,940 response. Data were fit to a nonlinear regression (four parameter model, GraphPad v. 8) for statistics in Table 1; or fit to the operational model to calculate bias (Fig. 9). Data are mean ± S.E.M. n ≥ 6 independent experiments performed in triplicate.
βarrestin2 recruitment
Seven of fifteen SCRAs displayed none or very weak (i.e. low efficacy and EC50 > 10,000 nM) agonist activity for the recruitment of βarrestin2 to CB2R relative to CP55,940 (Fig. 8, Table 1). ∆9-THC was assayed as a control partial agonist and displayed significantly lower efficacy, but similar potency, relative to CP55,940 (Fig. 8, Table 1). Four compounds: meta-AB-FUBINACA, ortho-AB-FUBINACA, 5-fluoro NPB-22, and 5-fluoro MPP-PICA were not different with regard to potency relative to the reference agonist CP55,940, and produced supramaximal EMax responses (i.e. > 100%) relative to CP55,940 (Fig. 8, Table 1). In contrast, five other compounds AM 2232, AB-PINACA N-(2-fluoropentyl) isomer, 3-chloro AB-PINACA, and 4-fluoro MDMB-BINACA displayed greater potency and produced supramaximal EMax responses (i.e. > 100%) relative to CP55,940 (Fig. 8, Table 1).

βarrestin2 recruitment to CB2R following SCRA treatment. CHO cells stably-expressing hCB2R were treated with 0.10 nM–10 μM SCRA 90 min and βarrestin2 recruitment was measured. βarrestin2 recruitment data are expressed as % CP55,940 response. Data were fit to a nonlinear regression (four parameter model, GraphPad v. 8) for statistics in Table 1; or fit to the operational model to calculate bias (Fig. 9). Data are mean ± S.E.M. n ≥ 6 independent experiments performed in triplicate. Note the difference in y-axis scale between a and all other panels.
Bias analyses
As with CB1R signaling, SCRAs were assessed for bias toward the inhibition of cAMP relative to βarrestin2 recruitment (i.e. ∆∆LogR > 0) at CB2R (Fig. 9). THC and three SCRAs—meta-AB-FUBINACA, 5-fluoro NPB-22, and 5-fluoro MPP-PICA did not display a bias for cAMP inhibition versus βarrestin2 recruitment (i.e. ∆∆LogR not different from 0) (Fig. 9). Three compounds were inactive in βarrestin2 and were therefore considered obligate cAMP-biased ligands: JWH 182, JWH 018 2′-napthyl-N-(3-methylbutyl) isomer, and MAM2201 N-(5-chloropentyl) analog (Fig. 9). Six compounds—ortho-AB-FUBINACA, AM 2232, FDU-NNEI, Cl 2201, JWH 145, and EG 018 displayed a significant bias toward the inhibition of cAMP versus recruitment of βarrestin2 (i.e. ∆∆LogR > 0) (Fig. 9) Finally, three compounds—AB-PINACA N-(2-fluoropentyl isomer, 3-chloro AB-PINACA, and 4-fluoro MDMB-BINACA—displayed a significant bias toward the recruitment of βarrestin2 relative to inhibition of FSK-stimulated cAMP (i.e. ∆∆LogR < 0) (Fig. 5).

CB2R SCRA bias. Compound bias between cAMP inhibition and βarrestin2 recruitment (ΔΔLogR (cAMP—βarr2)] is shown here for data presented in Figs. 7 and 8; Table 1. Data were fit to the operational model to calculate bias. Data are mean with 95% CI, n ≥ 6 independent experiments performed in triplicate. Values > 0 represent cAMP bias (white), values < 0 represent βarrestin2 bias (red), and values not different from 0 are unbiased (blue) as determined by 95% CI not overlap** with 0.
CB1R versus CB2R selectivity
With regard to receptor affinity, six SCRAs displayed greater affinity at CB1R relative to CB2R: JWH 018 2′-naphthyl-N-(3-methylbutyl) isomer, MAM2201 N-(5-chloropentyl) analog, AM 2232, FDU-NNEI, Cl 2201, and AB PINACA N-(2-fluoropentyl) isomer (Figs. 1, 6; Table 1). Interestingly, AB PINACA N-(2-fluoropentyl) isomer was unique in displaying a higher affinity for CB1R but only partially competing for [3H]CP55,940 binding at CB1R, whereas this SCRA fully competed with [3H]CP55,940 at CB2R (Figs. 1, 6; Table 1). Only one SCRA—4-fluoro MDMB-BINACA—displayed greater affinity at CB2R relative to CB1R (Figs. 1, 6; Table 1). FDU-NNEI displaced more [3H]CP55,940 at CB1R relative to CB2R (Figs. 1, 6; Table 1).
In the cAMP inhibition assay, several compounds displayed receptor subtype selectivity with regard to potency and efficacy. JWH 018 2′-naphthyl-N-(3-methylbutyl) isomer and AM 2232 were more potent at CB2R relative to CB1R; whereas MAM2201 N-(5-chloropnetyl) analog, 3-chloro AB-PINACA, and JWH 145 were more potent at CB1R relative to CB2R. JWH 182 displayed greater efficacy at CB2R than CB1R (Table 1).
In the βarrestin2 recruitment assay, meta-AB FUBINACA and 5-fluoro MPP-PICA were minimally active at CB1R but agonists at CB2R. Conversely, JWH 182 was inactive at CB2R but an agonist at CB1R (Table 1). Ortho-AB-FUBINACA, AB-PINACA N-(2-fluoropentyl) isomer, and 3-chloro AB-PINACA were more potent for CB2R-dependent recruitment of βarrestin2 than CB1R-dependent recruitment of βarrestin2 (Table 1). Eight SCRAs displayed significantly less efficacy for CB2R-dependent βarrestin2 recruitment relative to CB1R-dependent βarrestin2 recruitment, which is likely attributable to the supramaximal responses observed for several SCs at CB1R in this assay (Table 1).
In silico docking to CB1R and CB2R
Following our cell culture-based work, we wanted to determine whether the structures of these SCRAs might explain their ligand bias and receptor subtype selectivity. Binding of each SCRA to CB1R and CB2R was modelled via induced fit docking in Schrödinger using G protein-bound structures for each receptor (CB1R PDB 6N4B 20; CB2R PBD 6PT0 56,57.
Modified tetrad testing (catalepsy, body temperature, tail flick assay) was conducted according to previously described methods, which are summarized briefly here 19. Time points for testing were chosen based on previously published findings 19,22,23 and a time course experiment conducted with 10 mg/kg CP55,940 (Supplementary Fig. 3). Catalepsy was assessed in the ring holding assay 5 min following injection with mouse forepaws clasped to a 5 mm ring positioned 5 cm above the surface of the testing space. The length of time the ring was held was recorded (s) to a maximum time of 60 s (i.e. MPE = 60 s). Internal body temperature was measured 15 min after injection. Anti-nociception was determined by assessing tail flick latency 20 min after injection. Mouse tails were placed ~ 1 cm into 52 ± 2 °C water and the time until the animal removed its tail was recorded as tail flick latency (s) up to 20 s (i.e. MPE = 20 s).
EPM was conducted according to methods adapted from Marks et al. 58. The EPM was built from a plywood base painted blue with blue corrugated plastic floor and walls. The floor of the maze was composed of perpendicular interlocking arms (110 cm long × 10 cm wide) with two arms having 45 cm walls (closed arms) and two arms having no walls and a 0.5 cm edge (open arms), all elevated 45 cm from the ground 58. Trials began with the mouse in the central zone facing an open arm. Trials lasted 5 min and began 30 min after SCRA treatment. An arm entry was considered to be all four limbs leaving the central zone. Arm entries and the percent time spent in open versus closed arms were recorded by an observer blinded to the treatment group. Less time in the open arms and more time in the closed arms was used as a model of anxiogenic behaviour 58.
Statistical analyses
[3H]CP55,940 radioligand saturation binding data are provided as raw counts per minute (cpm) bound and fit to a one site total binding non-linear gression (GraphPad) (Supplementary Fig. 1). [3H]CP55,940 radioligand competition binding data are provided as % change from maximal 3H bound (i.e. 100%). Data for HitHunter cAMP and PathHunter βarrestin2 data are shown as % of maximal CP55,940 response (i.e. 100%). Estimates of Ki, EC50, Emin, and Emax were determined using non-linear regression with variable slope (four parameters) (GraphPad, Prism, v. 8.0). The operational model of Black and Leff 59 was used to estimate bias (∆∆LogR) with CP55,940 as the reference agonist 11,18,19. One-way analysis of variance (ANOVA), followed by Tukey’s post-hoc test, was used for statistical analyses (p < 0.05 determined to be significant); and Bartlett’s test was used to confirm homogeneity of variance (GraphPad). Values are presented as the mean ± the standard error of the mean (SEM) or 95% confidence interval (CI), as indicated in tables and figure legends.
References
Pertwee, R. G. Ligands that target cannabinoid receptors in the brain: From THC to anandamide and beyond. Addict. Biol. 13, 147–159 (2008).
Howlett, A. C. & Abood, M. E. CB1 and CB2 receptor pharmacology. Adv. Pharmacol. 80, 169–206 (2017).
Auwärter, V. et al. ‘Spice’ and other herbal belnds: Harmless incense or cannabinoid designer drugs?. J. Mass. Spectrom. 44, 832–837 (2009).
Banister, S. D. & Connor, M. The chemistry and pharmacology of synthetic cannabinoid receptor agaonist new psychoactive substances: Evolution. Handb. Exp. Pharmacol. 252, 191–226 (2018).
Labay, L. M. et al. Synthetic cannabinoid drug use as a cause or contributory cause of death. Forensic Sci. Int. 260, 31–39 (2016).
Hermanns-Clausen, M., Kneisel, S., Szabo, B. & Auwärter, V. Acute toxicity due to the confirmed consumption of synthetic cannabinoids: Clinical and laboratory findings. Addiction 108, 534–544 (2013).
Fantegrossi, W. E., Moran, J. H., Radominska-Pandya, A. & Prather, P. L. Distinct pharmacology and metabolism of K2 synthetic cannabinoids compared to ∆(9)-THC: Mechanism underlying greater toxicity?. Life Sci. 97, 45–54 (2014).
De Petrocellis, L., Ligresti, A. & Moriello, A. S. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 163, 1479–1494 (2011).
Sachdev, S. et al. In vitro determination of the efficacy of illicit synthetic cannabinoids at CB1 receptors. Br. J. Pharmacol. 176, 4653–4665 (2019).
Sachdev, S. et al. Differential activation of G protein-mediated signaling by synthetic cannabinoid receptor agonists. Pharmacol. Res. Perspect. 8, e00566 (2020).
Laprairie, R. B., Bagher, A. M., Kelly, M. E. & Denovan-Wright, E. M. Biased type 1 cannabinoid receptor signaling influences neuronal viability in a cell culture model of Huntington disease. Mol. Pharmacol. 89, 364–375 (2016).
Dhopeshwarkar, A. & Mackie, K. Functional selectivity of CB2 cannabinoid receptor ligands at a canonical and noncanonical pathway. J. Pharmacol. Exp. Ther. 358, 342–351 (2016).
Wouters, E., Walraed, J., Banister, S. D. & Stove, C. P. Insights into biased signaling at cannabinoid receptors: Synthetic cannabinoid receptor agonists. Biochem. Pharmacol. 169, 113623 (2019).
Banister, S. D. et al. Effects of bioisosteric fluorine in synthetic cannabinoid designer drugs JWH-018, AM-2201, UR-144, XLR-11, PB-22, 5F-PB-22, APICA, and STS-135. ACS Chem. Neurosci. 6, 1445–1458 (2015).
Banister, S. D. et al. Pharmacology of indole and indazole synthetic cannabinoid designer drugs AB-FUBINACA, ADB-FUBINACA, AB-PINACA, ADB-PINACA, 5F-AB-PINACA, 5F-ADB-PINACA, ADBICA, and 5F-ADBICA. ACS Chem. Neurosci. 6, 1546–1559 (2016).
Banister, S. D. et al. The chemistry and pharmacology of putative synthetic cannabinoid receptor agonist (SCRA) new psychoactive substances (NPS) 5F-PY-PICA, 5F-PY-PINACA, and their analogs. Drug. Test. Anal. 11, 976–989 (2019).
Patel, M. et al. Signalling profiles of a structurally diverse panel of synthetic cannabinoid receptor agonists. Biochem. Pharmacol. 175, 113871 (2020).
Laprairie, R. B. et al. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chem. Neurosci. 8, 1188–1203 (2017).
Zagzoog, A. et al. In vitro and in vivo pharmacological activity of minor cannabinoids isolated from Cannabis sativa. Sci. Rep. 10, 20405 (2020).
Krishna Kumar, K. et al. Structure of a signaling cannabinoid receptor 1-G protein complex. Cell 176, 448–458 (2019).
**ng, C. et al. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 180, 645–654 (2020).
Garai, S. et al. Application of fluorine- and nitrogen-walk approaches: defining the structural and functional diversity of 2-phenylindole class of cannabinoid 1 receptor positive allosteric modulators. J. Med. Chem. 63, 542–568 (2020).
Long, L. E. et al. A behavioural comparison of acute and chronic ∆9-tetrahydrocannabinol and cannabidiol in C57BL/6JArc mice. Int. J. Neuropsychopharmacol. 13, 861–876 (2010).
Li, X. et al. Crystal structure of the human cannabinoid receptor CB2. Cell 176, 459–467 (2019).
Hua, T. et al. Crystal structure of the human cannabinoid receptor CB1. Cell 167, 750–762 (2016).
Hua, T. et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547, 468–471 (2017).
Muller, C., Morales, P. & Reggio, P. H. Cannabinoid igands targeting TRP channels. Front. Mol. Neursoci. 15, 487 (2019).
Cannaert, A., Storme, J., Franz, F., Auwärter, V. & Stove, C. P. Detection and activity profiling of synthetic cannabinoids and their metabolites with a newly developed bioassay. Anal. Chem. 88, 11476–11485 (2016).
Noble, C., Cannaert, A., Linnet, K. & Stove, C. P. Application of an activity-based receptor bioassay to investigate the in vitro activity of selected indole- and indazole-3-carboxamide-based synthetic cannabinoids at CB1 and CB2 receptors. Drug Test. Anal. 11, 501–511 (2019).
Antonides, L. H. et al. Enantiospecific synthesis, chiral separation, and biological activity of four indazole-3-carboxamide-type synthetic cannabinoid receptor agonists and their detection in seized drug samples. Front. Chem. 7, 321 (2019).
Longworth, M. et al. Pharmacology of cumyl-carboxamide synthetic cannabinoid new psychoactive substances (NPS) CUMYL-BICA, CUMYL-PICA, CUMYL-5F-PICA, CUMYL-5F-PINACA, and their analogues. ACS Chem. Neurosci. 8, 2159–2167 (2017).
Longworth, M., Connor, M., Banister, S. D. & Kassiou, M. Synthesis and pharmacological profiling of the metabolites of synthetic cannabinoid drugs APICA, STS-135, ADB-PINACA, and 5F-ADB-PINACA. ACS Chem. Neurosci. 8, 1673–1680 (2017).
Wouters, E., Mogler, L., Cannaert, A., Auwärter, V. & Stove, C. Functional evaluation of carboxy metabolites of synthetic cannabinoid receptor agonists featuring scaffolds based on l-valine or l-tert-leucine. Drug Test. Anal. 11, 1183–1191 (2019).
Cawston, E. E. et al. Real-time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanisms of action. Br. J. Pharmacol. 170, 893–907 (2013).
Laprairie, R. B. et al. Probing the CB1 cannabinoid receptor binding pocket with AM6538, a high-affinity irreversible antagonist. Mol. Pharmacol. 96, 619–628 (2019).
Iliff, H. A., Lynch, D. A., Kotsikorou, E. & Reggio, P. H. Parameterization of Org27569: an allosteric modulator of the cannabinoid CB1 G protein-coupled receptor. J. Comput. Chem. 32, 2119–2126 (2011).
Bohn, L. M. et al. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498 (1999).
Schmid, C. L. et al. (2017) Bias factor and therapeutic window correlate to predict safe opioid analgesics. Cell 171, 1165–1175 (2017).
Wiley, J. L. et al. Cannabinoids in disguise: ∆9-tetrahydrocannabinol-like effects of tetramethylcyclopropyl ketone indoles. Neuropharmacol. 75, 145–154 (2013).
Kevin, R. C. et al. CUMYL-4CN-BINACA is an efficacious and potent pro-convulsant synthetic cannabinoid receptor agonist. Front. Pharmacol. 10, 595 (2019).
Wiley, J. L. et al. AB-CHMINACA, AB-PINACA, and FUBIMINA: Affinity and potency of novel synthetic cannabinoids in producing ∆9-tetrahydrocannabinol-like effects in mice. J. Pharmacol. Exp. Ther. 354, 328–339 (2015).
Ibsen, M. S. et al. Cannabinoid CB1 and CB2 receptor-mediated arrestin translocation: Species, subtype, and agonist-dependence. Front. Pharmacol. 10, 350 (2019).
Carlier, J. et al. Pharmacodynamic effects, pharmacokinetics, and metabolism of the synthetic cannabinoid AM-2201 in male rats. J. Pharmacol. Exp. Ther. 367, 543–550 (2018).
Adamowicz, P. Blood concentrations of synthetic cannabinoids. Clin. Toxicol. 59, 246–251 (2021).
Pike, E. et al. Systematic evaluation of a panel of 30 synthetic cannabinoid receptor agonists structurally related to MMB-4en-PICA, MDMB-4en-PINACA, ADB-4en-PINACA and MMB-4CN-BUTINACA using a combination of binding and different CB1 receptor activation assays PART I: Synthesis, analytical characterization, and binding affinity for human CB1 receptors. Drug Test Anal. https://doi.org/10.1002/dta.3037 (2021).
Grafinger, K. E. et al. Systematic evaluation of a panel of 30 synthetic cannabinoid receptor agonists structurally related to MMB-4en-PICA, MDMB-4en-PINACA, ADB-4en-PINACA and MMB-4CN-BUTINACA using a combination of binding and different CB 1 receptor activation assays PART II: Structure activity relationship assessment via a β-arrestin recruitment assay. Drug Test Anal. https://doi.org/10.1002/dta.3035 (2021).
Cannaert, A. et al. Synthesis and in vitro cannabinoid receptor 1 activity of recently detected synthetic cannabinoids 4F-MDMB-BICA, 5F-MPP-PICA, MMB-4en-PICA, CUMYL-CBMICA, ADB-BINACA, APP-BINACA, 4F-MDMB-BINACA, MDMB-4en-PINACA, A-CHMINACA, 5F-AB-P7AICA, 5F-MDMB-P7AICA, and 5F-AP7AICA. ACS Chem. Neurosci. 11, 4434–4446 (2020).
Frost, J. M. et al. Indol-3-yl-tetramethylcyclopropyl ketones: Effects of indole ring substitution on CB2 cannabinoid receptor activity. J. Med. Chem. 51, 1904–1912 (2008).
Wacker, D. et al. Crystal structure of an LSD-bound human serotonin receptor. Cell 168, 377–389 (2017).
Armenian, P. et al. Intoxication from the novel synthetic cannabinoids AB-PINACA and ADB-PINACA: A case series and review of the literature. Neuropharmacology 134, 82–91 (2018).
Bolognini, D. et al. The plant cannabinoid Δ9-tetrahydrocannabivarin can decrease signs of inflammation and inflammatory pain in mice. Br. J. Pharmacol. 160, 677–687 (2010).
Bolognini, D., Cascio, M. G., Parolaro, D. & Pertwee, R. G. AM630 behaves as a protean ligand at the human cannabinoid CB2 receptor. Br. J. Pharmacol. 165, 2561–2574 (2012).
Shao, Y. et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 8, 3172–3191 (2006).
Kuhn, B. et al. Prospective evaluation of free energy calculations for the prioritization of cathepsin L inhibitors. J. Med. Chem. 60, 2485–2497 (2017).
Hurst, D. P. et al. Identification of CB1 receptor allosteric sites using force-biased MMC simulated annealing and validation by structure-activity relationship studies. ACS Med. Chem. Lett. 10, 1216–1221 (2019).
Olfert, E. D. et al. (eds) Guide to the Care and Use of Experimental Animals 2nd Edn, Vol. 1. (Canadian Council on Animal Care, 2017).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 29, e1000412 (2010).
Marks, W. N. et al. The genetic absence epilepsy rats from Strasbourg model of absence epilepsy exhibits alterations in fear conditioning and latent inhibition consistent with psychiatric comorbidities in humans. Eur. J. Neurosci. 43, 25–40 (2016).
Black, J. W. & Leff, P. Operational models of pharmacological agonism. Proc. R. Soc. Lond. B. Biol. Sci. 220, 141–162 (1983).
Acknowledgements
Funding for this project was provided by a Health Canada Research Contract as well as a CIHR Partnership Grant with GlaxoSmithKline to RBL (#387577).
Author information
Authors and Affiliations
Contributions
A.Z. designed and conducted in vitro and in vivo experiments, analyzed data, and contributed to the editing of the manuscript; A.L.B. designed and conducted in silico experiments, analyzed data, and contributed to the editing of the manuscript; T.B. assisted with in vivo experiments; E.D.K. and R.B. assisted with in silico experiments and data analysis; M.P., Z.J., and M.N. assisted with experimental design, contributed to the editing of the manuscript, and provided access to critical materials; R.B.L. analyzed data, wrote and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zagzoog, A., Brandt, A.L., Black, T. et al. Assessment of select synthetic cannabinoid receptor agonist bias and selectivity between the type 1 and type 2 cannabinoid receptor. Sci Rep 11, 10611 (2021). https://doi.org/10.1038/s41598-021-90167-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90167-w
- Springer Nature Limited
This article is cited by
-
Cannabinoids in traumatic brain injury and related neuropathologies: preclinical and clinical research on endogenous, plant-derived, and synthetic compounds
Journal of Neuroinflammation (2023)
-
Simultaneous fatal poisoning of two victims with 4F-MDMB-BINACA and ethanol
Forensic Toxicology (2023)
-
Micro-paper-based analytical device decorated with metal-organic frameworks for the assay of synthetic cannabinoids in oral fluids coupled to ion mobility spectrometry
Microchimica Acta (2023)