
Abstract
To better predict population evolution of invasive species in introduced areas it is critical to identify and understand the mechanisms driving genetic diversity and structure in their native range. Here, we combined analyses of the mitochondrial COI gene and 11 microsatellite markers to investigate both past demographic history and contemporaneous genetic structure in the native area of the gastropod Tritia neritea, using Bayesian skyline plots (BSP), multivariate analyses and Bayesian clustering. The BSP framework revealed population expansions, dated after the last glacial maximum. The haplotype network revealed a strong geographic clustering. Multivariate analyses and Bayesian clustering highlighted the strong genetic structure at all scales, between the Black Sea and the Adriatic Sea, but also within basins. Within basins, a random pattern of genetic patchiness was observed, suggesting a superimposition of processes involving natural biological effects (no larval phase and thus limited larval dispersal) and putative anthropogenic transport of specimens. Contrary to the introduced area, no isolation-by-distance patterns were recovered in the Mediterranean or the Black Seas, highlighting different mechanisms at play on both native and introduced areas, triggering unknown consequences for species’ evolutionary trajectories. These results of Tritia neritea populations on its native range highlight a mixture of ancient and recent processes, with the effects of paleoclimates and life history traits likely tangled with the effects of human-mediated dispersal.
Similar content being viewed by others

Introduction
The description of mechanisms driving genetic diversity and structure of invasive species in their native range has proven to be of considerable use to predict further evolution in their introduced areas1,2,3,4. However, it is always difficult to disentangle the natural effects of life history traits and historical events (such as past climate changes) over species geographic ranges from recent human-related range expansion5. This is even more true for species whose introduced area is close to the native range, and only genetic tools can discriminate between a natural spread or a human-mediated introduction6,7,8,9. Life history traits are considered to be major drivers of population genetic diversity and structure10. In particular, species with direct development (no planktonic stage) have reduced potential for dispersal and usually show strong genetic structure11,12,13. The effects of past climate changes on population genetic structure and distribution have also been extensively reported14,15. For instance, sea-level changes in the Pleistocene often led to population fragmentations thus creating a dynamic of expansion/contraction of populations over time16. In the last decades, cases of human-related range expansion have increasingly been reported and the natural patterns of biodiversity have been altered by artificial translocation patterns17,18,19. The superimposition of these various evolutionary processes makes it difficult to evaluate the potential outcome of the future of introduced species.
Tritia neritea (Linnaeus, 1758), a scavenging nassariid gastropod, is a direct developer, with juveniles hatching as crawl-aways from single-embryo capsules attached to hard substrates20,21. This species was long known in the literature as Cyclope neritea, but the former genus Cyclope was recently moved to Tritia based on genetic results22. Two living species are currently recognized in the study region23,24,25, i.e. T. neritea (Linnaeus, 1758) and T. pellucida (Risso, 1826). Tritia neritea is euryhaline, found in lagoonal and river-influenced sandy and muddy bottoms26, which results in discontinuities of populations, with high densities in lagoons, bays, estuaries27,28,29,30. Its native range spreads across the Mediterranean, Black and Azov Seas to the adjacent eastern Atlantic coasts of Morocco and south Iberian Peninsula24,31,32,33,34,35. Although presenting a patchy distribution as well36, the case of the Black Sea is somewhat different given that its average salinity ranges between 18 and 22 psu, suitable to euryhaline species such as T. neritea.
The evolutionary history of the species and genus is complex. According to Gili & Martinell37, Tritia neritea originates from an early Pliocene ancestor identified as Cyclope migliorinii (Bevilacqua, 1928). Because of the basin-wide geographic range of T. migliorinii, it is impossible to formulate a robust hypothesis about the center of origin of T. neritea. Tritia migliorinii was characterized by a planktotrophic development and went extinct in the Pleistocene whereas T. neritea, lacking a larval phase, survived37. Such Pliocene-planktotrophic/Recent-non-planktotrophic pairs are common among gastropods of the Mediterranean while the reason for such trend is not yet clearly understood38. Climate changes strongly influenced the evolutionary and biogeographic patterns of marine organisms, with sea-level fluctuations triggering isolation of populations. In particular, for the Mediterranean Sea, Plio-Pleistocene sea-level changes often led to population fragmentations sha** species distributions and population genetic structures of many fish or marine invertebrates39,40,41,42. However, in the specific case of euryhaline gastropods, such as T. neritea, this process is not so obvious. On one hand, the populations restricted to lagoons are, in theory, exposed to habitat loss during sea-level changes, thus compromising population survival. In contrast, populations that inhabit coastal nearshore bottoms may prove to be more resilient to disconnection of their habitat during sea-level fluctuations.
The natural history background of Tritia neritea clearly depicts a rather complex situation with respect to population connectivity through space and time. Furthermore, starting from the 1970s, T. neritea was recorded further North, in Galicia43, in the Bay of Biscay34,44,45 and up to the English Channel46. Mitochondrial sequence data demonstrated the introduction in these areas45,46,47 (as opposed to a natural spread of the species). However, the history of introduction seems to be complex, with multiple independent introduction events from several sources. For instance, the North Adriatic region was proposed as a source for the North Western Iberian Peninsula invasion with the Manila clam, Ruditapes philippinarum, trade as the likely vector47, whereas the North Western Mediterranean and Portuguese areas were pointed as sources for the Bay of Biscay’s invasive population with the oyster trade as vector45. These and other studies on T. neritea focused on the introduced areas and included only one to few localities within the native range. Only Simon-Bouhet et al.46 included multiple localities in the native range, however, they only analyzed mitochondrial markers. While microsatellite markers were developed48, they have never been used to further our collective understanding of the population genetic structure of T. neritea. To add further complexity to such an intricate distribution, some populations are thought to represent recent introduction events within the ‘native’ range, such is likely the case for the Gulf of Gabès49. Consequently, it may prove very challenging to track unequivocally clear dispersal pathways (natural and/or artificial) for this species, and a better understanding of the population structure in the native area is critical.
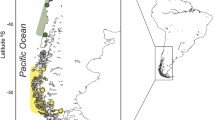
Herein, we investigate the genetic structure and demographic history of Tritia neritea within its native range throughout the Mediterranean and the Black Seas (Fig. 1; Table 1). We aim to decipher biogeographic patterns considering the geological and climatic history of the region, with multiple disconnections of the Black Sea and the Adriatic Sea from the main Mediterranean region during low sea levels and subsequent recolonization following sea level rises50,51,52,53. In particular, population genetic structures of species from the Black Sea have seldom been assessed, as well as their relation to other populations in the Mediterranean Sea. However, a recent study on the black scorpionfish Scorpaena porcus clearly showed restricted gene flow between these two basins and demographic expansions of populations dated after the last glacial maximum. Therefore, the combination of the past history of the Mediterranean Sea, the patchy habitat distribution of the species and the low potential for dispersal resulting from the reproductive mode are likely to have shaped the evolutionary history of T. neritea and resulted in strong population structures at all scales. To test this, our study combines analyses of the mitochondrial COI gene and 11 microsatellite markers to investigate both past demographic history and contemporary genetic structure within the native geographic distribution of T. neritea, using coalescent theory and Bayesian skyline reconstruction, multivariate analyses and Bayesian clustering.

Map of the 17 localities sampled for Tritia neritea in the Mediterranean Sea.
Results
Mitochondrial data
A 524 bp portion of the COI gene was sequenced for 128 specimens, resulting in 20 haplotypes; 12 of which are new (Table 2). A strong geographic clustering is noticeable on the haplotype network colored by basins with 5 haplogroups visible and a central group (Fig. 2). Interestingly, the central group of haplotypes was only recorded in the invaded area and their source is unknown51. Samples from the Aegean Sea, the Black Sea and the South European Atlantic Shelf / Western Mediterranean Sea each form a distinct cluster of haplotypes. Samples from the Adriatic Sea are spread in the 2 remaining haplogroups, one with only Adriatic samples and the other one with samples from the Gulf of Lion (Western Mediterranean Sea). Noticeably, like most of the sequences from the west Mediterranean Sea already published (263 specimens), our 23 sequences from Fusaro lake and Cape Bon (Tunis) correspond to haplotype H1.

Median joining network of haplotypes of the 128 COI sequences of Tritia neritea generated for this study together with haplotypes available in GenBank. Circle sizes are proportional to the number of sequences per haplotype. Distances are proportional to the number of mutations between haplotypes. Numbers in red indicate numbers of mutations between haplotypes. The haplotypes marked as unknown origin were sampled in an introduced area, the Bay of Biscay46, but the population of origin in the native area is unknown.
The Bayesian skyline plot did not show a population expansion based on only our samples from the Black Sea. However, a population expansion was detected for samples from the Adriatic Sea (Fig. 3A). When considering additional sequences available on GenBank, an expansion of population can also be distinguished for the Black Sea (Fig. 3B) and the signal of expansion in the Adriatic Sea becomes stronger (steeper slope in Fig. 3B). According to the molecular clock used, the age of expansion is datable at around 10,000–15,000 years ago, i.e. after the Last Glacial Maximum, for the Adriatic Sea population and around 5000–10,000 years ago for the Black Sea population.

Bayesian Skyline Plots of Tritia neritea specimens from the Adriatic Sea and the Black Sea reconstructed from (A) sequences from this study only and (B) together with sequences already published. The X-axis indicates the time in years; the Y-axis indicates the female effective population size (NeT, with T = generation time). The black line is the median estimate of the estimated effective population size. The two blue lines are the upper and the lower estimates of 95% interval.
Microsatellite data
A total of 786 specimens were analyzed at 11 loci. Heterozygosity values (Ho) ranged from 0.356 to 0.567 and mean allele numbers ranged from 5.09 to 11.55 (Table 1). Fis values were high and significant for all populations sampled, ranging from 12 to 33% (Table 1), and this signal was not due to a single or few loci (data not shown). All pairwise Fst values were significantly different from zero, except those comparing Ancona and Venice in the northern Adriatic Sea, and Costinesti2 and CostinestiB in the Black Sea (Table 3).
Among basins genetic differentiation
The PCoA analysis showed the grou** of samples from the Western Mediterranean (Fusaro and Tunis) with samples from the Aegean Sea (Paralia Katerini) and samples from the Adriatic Sea (Fig. 4). Samples from the Black Sea clustered together. The Structure analysis on the total dataset revealed K = 2 groups as the most likely partitioning (Fig. S1). Using the sampling localities as prior (ocean basin) did not change the results (data not shown). Noticeably, the Western Mediterranean and the Aegean Sea samples clustered with samples from the Adriatic Sea and samples from the Black Sea form another cluster (Fig. 5). The sample from Sile shows a mixed genetic make-up, with some individuals presenting a proportion of their genome with an Adriatic Sea affinity.

Principal Coordinates Analysis (PCoA) of the 17 localities sampled for Tritia neritea genotyped at 11 microsatellite markers. Coordinate 1 explains 40% of the variation while coordinate 2 explains 15% of the variation.

Bayesian clustering analysis showing the most likely partition for: (A) the total 786 multi-locus genotypes of Tritia neritea analyzed in this study; (B) the Adriatic samples only; (C) the Black Sea samples only, without Ropotamo.
Within basins genetic differentiation
For T. neritea in the Adriatic Sea, all the Fst values were significant, except for the Venice-Ancona pairwise comparison. The Structure plot revealed the most likely partitioning to be of K = 2 groups (Fig. 5B) corresponding to a first group composed of the northern localities (Ancona + Venice + Nin) together with Otranto in the south; and a second group with the southern localities (Karaburun, Porto Cesareo, Torre Guaceto, Amvrakikos). This grou** was also distinguishable on the PCoA (Supplementary Fig. S2). For T. neritea within the Black Sea, all the Fst values were significant. The Structure plot revealed K = 2 clusters to be most likely. The groups were (i) Costinesti2, Kaliakra1, Kaliakra2, CostinestiB; (ii) CostinestiA, Sile and Karadag, Tarkhankut; such as also separated on the PCoA (Fig. 4, Supplementary Fig. S1). Furthermore, the Mantel tests did not reveal any pattern of isolation by distance for both the Adriatic Sea (Z statistic = 6774, Pearson’s r = 0.09, p value > 0.05) and the Black Sea (Z statistic = 8629, Pearson’s r = 0.315, p value > 0.05) basins.
Parentage analysis
A total of 82 fullsib and 206 halfsib relationships were recovered among the 786 multilocus genotypes (Table S2). Most of the relationships occur within localities or close-by localities.
Discussion
As expected for species characterized by a direct cycle without a pelagic larval stage, we observed genetically divergent populations even at local scales within small seas such as the Adriatic Sea or the Black Sea. The close investigation of this species within its native range demonstrates complex diversity patterns, genetic structure and historical changes that improve our understanding of patterns in the current and future introduced areas.
The mitochondrial dataset confirms a strong geographic clustering of haplotypes (Fig. 2), as already noted in46,54,55. Five mitochondrial clades correspond to marine ecoregions as defined in56: (i) South European Atlantic shelf and Western Mediterranean, (ii) Adriatic Sea, (iii) Aegean Sea and (iv) the Black Sea and (v) the 5th one is composed of specimens from both the Adriatic Sea and the Western Mediterranean Sea (Gulf of Lion). Noticeably, a group of haplotypes lies in the middle of the network, but was only recorded in the invaded area (North-Eastern Atlantic) while their source in the Mediterranean Sea remains unknown54. This group likely comes from an unsampled area of the native range (such as the South Eastern Mediterranean and Far Eastern Mediterranean basins) and its central position points toward an ancestral nature. As previously mentioned, if T. neritea originates from the widespread T. migliorinii, it is difficult to formulate a robust hypothesis about its center of origin. However, Gili & Martinell37 postulated a derivation of ‘Cyclope’ from a Nassarius (Plicarcularia) group containing two extant Mediterranean species (Tritia gibbosula and T. circumcincta), with a Levantine/southern Mediterranean distribution. Additionally, based on an east–west genetic diversity gradient, Simon-Bouhet46 hypothesized an eastern origin for T. neritea. Accordingly, our sample from the Aegean Sea demonstrates a high number of alleles and a high number of private alleles at nuclear loci. Further sampling on the South Eastern and Far Eastern Mediterranean coasts is warranted to locate and clarify the origin of these so far unsampled native haplotypes.
Whatever the origin of the species, the five mitochondrial lineages corresponding to distinct marine basins were likely differentiated during paleoclimate sea-level fluctuations. Simon-Bouhet46 hypothesized the persistence of T. neritea populations in several distinct refugia during glacial periods. Several refugia have been well documented in the Atlanto-Mediterranean basin41,42,57 corresponding to the Iberian Peninsula, the Western Mediterranean, the Eastern Mediterranean, etc. Based upon its current geographic distribution, T. neritea appears to be a temperate marine taxon. It might have responded to the lowered sea-surface temperature at glacial times, which favored the basinal spread of boreo-celtic species58, by shrinking its geographic range in the Mediterranean Sea to milder sectors of the basin46, e.g. the Levantine region. It is thus conceivable that we had geographically-distant stocks at the peak of the last glacial period in a disconnected mosaic-like situation. The post-glacial warming may have allowed for the progressive recolonization of sectors of the basin, resulting in the present pan-Mediterranean distribution. The high private allele number in Amvrakikos and Karaburun supports this hypothesis. Specimens from a refugium located in the Ionian Sea might have recolonized the Adriatic Sea, as seen in the decapod Carcinus aestuarii59. Likewise, the high private allele number at the Aegean location suggests a refugial area.
Therefore, our dataset seems consistent with the hypothesis that past climate changes impacted the genetic structure of T. neritea. Furthermore, the Bayesian skyline plots revealed population expansions in both seas when enough sequences are available (Fig. 3). Interestingly, the signal of population expansion is detectable even in the face of the strong genetic structure of T. neritea, a feature known to potentially hinder the detection of demographic expansion60. This expansion is dated from just after the Last Glacial Maximum for the Adriatic Sea population, as also seen for other marine invertebrates and fish in the region53,61 including other nassariid gastropod species62. In the specific case of the Black Sea, this basin achieved its current marine status only ca. 7000 years ago63. Before its post-glacial inundation, it was ascertained that the Black Sea, together with the Marmara Sea, was a lacustrine basin (Neoeuxinian stage) completely secluded from the Mediterranean Sea63,64. This fact rules out any chance that any marine refuges existed (even if euryhaline) within the Black Sea during the last glaciation, a theoretical claim put forth for other organisms but not applicable to T. neritea. In agreement with this age, fossil evidence from the Kerch Strait supports the occurrence of T. neritea in the Late Holocene65, thus setting a tempo for the Black Sea population presence in the basin. The settling of the modern Black Sea by T. neritea is thus a geologically-young phenomenon (< 7,000 years ago) that took place from the Aegean Sea via the Marmara Sea. Interestingly, the expansion of population for the Black Sea is dated at 5000–10,000 ya, which would agree with the timeframe of the recolonization of this sea proposed in the literature.
Regarding genetic diversity, overall, the heterozygosity at nuclear loci of Tritia neritea in its native range (0.486 < He < 0.724, Table 1) is comparable to that of other gastropods66,67,68, although lower than the values for another direct develo** gastropod species Crepidula convexa52 (0.885 < He < 0.943) or the broadcast spawner C. fornicata69 (0.806 < He < 0.838). Additionally, all the Fis values are high and significant, demonstrating a heterozygote deficiency in every populations. Such high values are not surprising for a species with a lack of planktonic larval phase, thus prone to inbreeding50 but were also detected for the mussel Mytilus galloprovincialis, a species with long-lived pelagic larvae70. Heterozygote deficiency is a common phenomenon in marine molluscs71. Some of the proposed explanations to this phenomenon are null alleles, local inbreeding, selection against heterozygotes and life history traits that produce spatial or temporal population substructure leading to a Wahlund effects70,71,72,73. Parentage analysis (Table S2) revealed half-sib (206 pairs) and full-sib (82 pairs) relationships within localities or between very close localities, and family structure could thus explain some of the heterozygote deficiencies detected. With a low dispersal potential for T. neritea, a sample likely comprises multiple related individuals, but also multiple families, which would trigger a high genetic diversity together with significant Fis values. Such Wahlund effect on multiple families with different allelic frequencies has been suggested for the Siphonaria limpets72. Other direct developer gastropods such as Crepidula convexa in the USA also display high and significant values of Fis51,52. Other direct developer marine invertebrates in the Mediterranean Sea also show high levels of inbreeding12,74,75.
Regarding genetic differentiation between basins, the pairwise Fst values, PCoA and Bayesian clustering all showed genetic differentiation between the Black Sea and the Mediterranean Sea populations, with the Aegean Sea and the Western Mediterranean populations clustering with the Adriatic population. There are also more private alleles in the Adriatic Sea than in the Black Sea. Genetic diversity and total number of alleles are also lower in the Black Sea compared to the Adriatic Sea (in spite of a higher number of specimens collected in the Black Sea), as previously noticed for several species of fish53,76,77. Only few studies have investigated population genetics for species distributed both in the Black Sea and the rest of the Mediterranean Sea, however a genetic differentiation appears to be the trend for the fish populations studied so far53,78,79,80,81. Furthermore, the mussel Mytilus galloprovincialis also showed strong genetic differentiation between its populations of the Black Sea and the rest of the Mediterranean Sea70. Some explanations for these differentiations can be found in the oceanographic currents, the presence of narrow straits (e.g. Bosphorus) and specific environmental conditions (in particular, low salinity) of the Black Sea80,81. Additionally, the Otranto Strait and the Aegean Front have been demonstrated to influence population genetic structures, however to a lesser extent for species without long pelagic larval phases compared to species with long pelagic larval phases82. Noticeably, the relatedness of individuals within basins could also trigger some proportion of the differentiation detected, as family groups are known to influence the inferences made with the program Structure83,84. However, recent literature advises against purging related individuals in population genetics85, since the removal of individuals also reduces precision of genetic estimates. This holds particularly true when errors in pedigree reconstructions are expected, as in our case due to the limited number of loci. Finally, discrepancies between the Atlantic and the Mediterranean population structures also exist. In the Mediterranean, we did not find a signal of isolation-by-distance, contrary to what was shown in the Atlantic population of T. neritea using the COI mitochondrial gene46,47,55 . This discrepancy might be linked to the very distinct mechanisms at play during introductions on new areas and natural evolutionary history on the native areas. The introduced area was likely home to a linear spread along the Atlantic shore over a short period of time, with founding events typical of colonization of new areas47, whereas in the native area, the strong genetic structure and chaotic pattern are likely due to rare long-distance dispersal events in the Mediterranean Sea over evolutionary periods and relicts of glacial disconnected refuges.
Within basins, strong genetic structures were also detectable, however they were “unpatterned” or “random” with no clear geographic clustering. Importantly, the half-sib and full-sib relationships detected within and among localities might increase the number of detected clusters83, suggesting that these results should be taken with caution. The Adriatic Sea did not show an east/west genetic differentiation, contrary to a recent study on the scorpionfish, Scorpaena porcus, sampled at the same localities53. Here, a north/south differentiation seems to arise, however, not perfect, as the Otranto sample clusters with the northern localities and the Amvrakikos sample comprises individuals with genetic make-up of the northern cluster. Within the Black Sea, spatially close samples such as Costinesti A and B or Kaliakra 1 and 2 also show genetic differentiation. Life history traits, such as local vs. broadcast dispersal, are known to have strong effects on genetic structure10,11 and the strong genetic differentiation at local scales is thus not surprising for T. neritea, a species without a dispersal stage. The Polyplacophora Lepidopleurus cajetanus, with a low potential for dispersal (lecithotrophic larvae) also exhibited a ‘chaotic patchiness’ pattern in the Mediterranean basin defined by a high genetic variability with locality-exclusive haplotypes, high genetic divergence, and a lack of geographic structure86. The term “chaotic genetic patchiness” was defined for species with a potential for dispersal and that show strong genetic structure at scales smaller than their dispersal potential and where “instantaneous drift” between cohorts is common87. Tritia neritea has no larval dispersal phase but seems to be especially prone to demonstrate such “random” genetic patchiness. The mechanisms underlying this pattern remain unknown but are likely a mixture of ‘natural’ and ‘artificial’ processes. For direct developers, rafting on floating objects has been proposed as a surrogate88,89, and would likely be sporadic, not recurrent, and not linked to geography, thus responsible for creating this kind of genetic patchiness. Oyster culture is also a vector for the introduction of Tritia neritea on the Atlantic coast45 and populations could be moved similarly within the native range (see below).
Samples from Sile demonstrated a mixed genetic make-up at the nuclear loci compared to the remaining of the Black Sea populations. The mitochondrial sequences, however, belong to the Black Sea haplogroup. Similarly, Simon-Bouhet54, based on mito-nuclear discordance raised the possibility of the introduced nature of a population at Mar Menor (Western Mediterranean Sea) and Aissaoui et al.49 documented a recent introduction event in the Gulf of Gabès (Tunisia), both populations are located in the native area. Likewise, the mixed genetic make-up of our sample from Sile, might be due to recent introductions of Mediterranean specimens into the Black Sea. Sile has a marina for yachts and small to medium sized fishing boats, which make seasonal trips to the Marmara and Aegean Seas. The discrepancy between nuclear and mitochondrial markers could be explained by mitochondrial capture (i.e. complete introgression), a phenomenon known in many organisms90,91,92 and mitochondrial introgression has been reported for mollusks93,94. Alternatively, if only male specimens were introduced, the nuclear dataset would be of foreign origin while the mitochondrial marker would remain from the geographic location sampled. However, no data on sex ratio is available for T. neritea to support this second hypothesis. Further study is warranted to verify if this population shows any effects of admixture (i.e. mixing of specimens from different genetic clusters), such as hybrid vigor. Rius & Darling95 recently highlighted the unknown evolutionary trajectory for such admixed populations. Human activities are responsible for the translocation of vast amounts of organisms, altering natural patterns of dispersal and gene flow. Anthropogenic effects on the native area have been demonstrated in the ascidian Ciona intestinalis that displayed genetic homogeneity among both close and distant sites and dissimilar genetic composition between close sites5. Similarly, a recent study on hydroids revealed contrasted patterns of strong genetic structure on local and regional scales contrasting with some haplotypes shared among ocean basins, revealing the likely effect of human-mediated transport96.
Conclusion
The patterns seen from our samples of Tritia neritea on its native range seem to be a mixture of recent and ancient processes, with the effects of paleoclimates and life history traits likely tangled with the effects of human-mediated dispersal. As anthropogenic pressure grows, it is going to be more and more difficult to disentangle the natural and artificial patterns of biodiversity.
Methods
Samples
In this study, through the application of the sampling scheme of the CoCoNet project97, a total of 786 specimens from 17 localities were analyzed (1 locality from the Aegean Sea, 2 localities from the West Mediterranean Sea, 8 localities from the Adriatic Sea + Ionian Sea (herein the 3 localities in the Ionian Sea close to the Adriatic Sea will be broadly included in the Adriatic Sea localities) and 6 localities from the Black Sea, see Fig. 1 and Table 1 for details). Samples were collected from the infralittoral by snorkeling or diving at a depth between 1 and 5 m, in 2013–2014. DNA was extracted from foot muscle using the PureGene protocol with a QIAxtractor robot (QiaGen, Hilden, Germany).
Molecular analyses
Mitochondrial sequences
A portion of the Cytochrome oxidase subunit 1 (COI) was amplified for a subset of 128 specimens including all locations, using the specific primers Cy2 5′-GTTAAAATTTCGATCTGTTA-3′ and CyR 5′-GGATTAGTTGGTACAGC-3′45. PCR mixture and cycling parameters were as given in this publication. The 42 haplotypes available from GenBank45,46,47,55 together with their frequencies were added to this newly generated dataset in order to place our samples in a broader context. Notably, a group of these haplotypes lies in the middle of the haplotype network, but have been only recorded in the invaded area and their source remains unknown54.
Microsatellite markers
A set of 14 microsatellite markers were screened for this study, 7 markers are from Simon-Bouhet et al.48 and 7 come from a new microsatellite library (Genoscreen, Lille, France) generated specifically for this study (see Supplementary Table S1 for locus details). PCR were performed in 10 µl volume containing: 5 µL of QIAGEN Multiplex PCR master mix, 3 µL of RNase free water (provided with the QIAGEN type-it Multiplex PCR Master Mix), 0.02 µl of each primer (100 µM) and 1 µl of DNA template. Three multiplexes of four, five and five loci, respectively, were run for each specimen. Cycling parameters were as follows: 15 min at 95 °C followed by 30 cycles of: (i) 30 s at 94 °C, (ii) 1 min 30 s at the optimal annealing temperature (55 °C, 57 °C or 60 °C) and (iii) 1 min at 72 °C and a final step at 57 °C during 30 min. Amplifications were verified on 1% Agarose gel. PCR products were sent to a private company for genoty** (GenoScreen, Lille, France). Genotypes were scored using GeneMapper v4.0 (Applied Biosystems).
Data analyses
Mitochondrial sequences
COI sequences were aligned using Mafft v.7 online98. Haplotype and nucleotide diversity indices as well as neutrality test statistics were computed in DnaSP v.599. Haplotype networks were computed with Network v5.0.0.3 (www. fluxus.engineering.com) using the Median Joining algorithm100. Mr AIC101 was used to determine the best fit model of nucleotide substitutions. Changes in population sizes were investigated using the Bayesian skyline plot (BSP) framework in Beast 2102. As the assumptions of this analysis framework are that the sequences represent a small sample from a haploid population evolving under Wright–Fisher dynamics, the BSPs were computed for each basin separately (Adriatic Sea / Black Sea) in order to try and work on samples that are closer to a single panmictic population. A total of 10,000,000 generations sampled every 100th generation were run. As no mutation rate is calibrated for Tritia neritea, a mean mutation rate ranging from 0.012 to 0.016 (substitutions per site per My), used for the COI gene in Protostomia103 and in limpets104 was applied.
Microsatellite markers
The presence of null alleles, scoring errors or large allele drop-out were verified using micro-checker105. As null alleles are common in mollusks106, we decided to discard the three microsatellite markers that presented an estimated frequency of null alleles above 0.10. Further analyses were run on the dataset with 11 loci. Summary statistics were computed in Genetix v.4.05.02107: the mean number of alleles (A), the expected (He) and observed (Ho) heterozygosities, the inbreeding coefficient (Fis) and the fixation index (Fst). The correction for multiple comparisons of Benjamini & Hochberg108 was applied to the Fst p values. A Principal Coordinate Analysis (PCoA) was computed in Genalex v6.5109. The most likely number of clusters present in our dataset was estimated using Structure v2.3110. This analysis was run both with and without prior on the location. After several first runs, the parameters were set as follows: a burn-in period of 100,000 iterations followed by 500,000 recorded iterations for K = 1 to K = 8 clusters and 15 iterations per K values. The most probable number of clusters present in this dataset was determined using the Evanno’s ΔK approach111 using Structure Harvester online112. Finally, a pattern of Isolation-by-distance was tested by a Mantel test in Genetix using the genetic distance [Fst/(1 − Fst)]113 and the log coastal geographical distance (in km) between each pair of localities within basins. Significance was obtained using a random permutation procedure implemented in Genetix (5,000 permutations). Finally, a parentage analysis was run using the software Colony114. Even if the number of loci is low for this kind of analyses84, and given that heterozygote deficiency is often detected in mollusk population genetic analyses71, we wanted to verify any sign of family structure in our dataset. This relatedness analysis was performed on the whole dataset including the 786 multilocus genotypes and with the following input parameters: a mating system with female and male polygamy, with possible inbreeding and without clones. The method was composed of 3 runs set as ‘long’, with a full likelihood method and a likelihood precision that was set to ‘high’.
Data availability
Sequences are available in GenBank (accession numbers: MN577950–MN577969).
References
Carlton, J. T. Pattern, process, and prediction in marine invasion ecology. Biol. Conserv. 78, 97–106. https://doi.org/10.1016/0006-3207(96)00020-1 (1996).
Stepien, C. A., Brown, J. E., Neilson, M. E. & Tumeo, M. A. Genetic diversity of invasive species in the Great Lakes versus their Eurasian source populations: insights for risk analysis. Risk Anal. 25, 1043–1060. https://doi.org/10.1111/j.1539-6924.2005.00655.x (2005).
Geller, J. B., Darling, J. A. & Carlton, J. T. Genetic perspectives on marine biological invasions. Annu. Rev. Mar. Sci. 2, 367–393. https://doi.org/10.1146/annurev.marine.010908.163745 (2010).
Estoup, A. & Guillemaud, T. Reconstructing routes of invasion using genetic data: why, how and so what?. Mol. Ecol. 19, 4113–4130. https://doi.org/10.1111/j.1365-294X.2010.04773.x (2010).
Hudson, J., Viard, F., Roby, C. & Rius, M. Anthropogenic transport of species across native ranges: unpredictable genetic and evolutionary consequences. Biol. Lett. https://doi.org/10.1098/rsbl.2016.0620 (2016).
Carlton, J. T. Biological invasions and cryptogenic species. Ecology 77, 1653–1655. https://doi.org/10.2307/2265767 (1996).
Holland, B. S. Genetics of marine bioinvasions. Hydrobiologia 420, 63–71. https://doi.org/10.1023/a:1003929519809 (2000).
Reitzel, A. M., Herrera, S., Layden, M. J., Martindale, M. Q. & Shank, T. M. Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol. Ecol. 22, 2953–2970. https://doi.org/10.1111/mec.12228 (2013).
Darling, J. A. et al. Recommendations for develo** and applying genetic tools to assess and manage biological invasions in marine ecosystems. Mar. Pol. 85, 54–64. https://doi.org/10.1016/j.marpol.2017.08.014 (2017).
Palumbi, S. R. Genetic-divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 25, 547–572. https://doi.org/10.1146/annurev.ecolsys.25.1.547 (1994).
Kelly, R. P. & Palumbi, S. R. Genetic Structure among 50 species of the northeastern Pacific rocky intertidal community. PLoS ONE 5, 13. https://doi.org/10.1371/journal.pone.0008594 (2010).
Boissin, E., Stohr, S. & Chenuil, A. Did vicariance and adaptation drive cryptic speciation and evolution of brooding in Ophioderma longicauda (Echinodermata: Ophiuroidea), a common Atlanto-Mediterranean ophiuroid?. Mol. Ecol. 20, 4737–4755. https://doi.org/10.1111/j.1365-294X.2011.05309.x (2011).
Selkoe, K. A. & Toonen, R. J. Marine connectivity: a new look at pelagic larval duration and genetic metrics of dispersal. Mar. Ecol. Prog. Ser. 436, 291–305. https://doi.org/10.3354/meps09238 (2011).
Stewart, J. R. & Lister, A. M. Cryptic northern refugia and the origins of the modern biota. Trends Ecol. Evol. 16, 608–613. https://doi.org/10.1016/s0169-5347(01)02338-2 (2001).
Hewitt, G. M. Genetic consequences of climatic oscillations in the quaternary. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 359, 183–195. https://doi.org/10.1098/rstb.2003.1388 (2004).
Provan, J. & Bennett, K. D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23, 564–571. https://doi.org/10.1016/j.tree.2008.06.010 (2008).
Carlton, J. T. & Geller, J. B. Ecological roulette—the global transport of nonindigenous marine organisms. Science 261, 78–82. https://doi.org/10.1126/science.261.5117.78 (1993).
Ruiz, G. M., Fofonoff, P. W., Carlton, J. T., Wonham, M. J. & Hines, A. H. Invasion of coastal marine communities in North America: apparent patterns, processes, and biases. Annu. Rev. Ecol. Syst. 31, 481–531. https://doi.org/10.1146/annurev.ecolsys.31.1.481 (2000).
Molnar, J. L., Gamboa, R. L., Revenga, C. & Spalding, M. D. Assessing the global threat of invasive species to marine biodiversity. Front. Ecol. Environ. 6, 485–492. https://doi.org/10.1890/070064 (2008).
Morton, J. The habits of Cyclope neritea, a style-bearing stenoglossan gastropod. Proc. Malacol. Soc. Lond. 34, 96–105 (1960).
Gomoiu, M. T. Biologisches Studium der Arten Nassa reticulata L. und Cyclonassa neritea (L.) im Schwarzen Meer (rumänischer Küstenbereich). Rev. Roum. Biol. Ser. Zool. 9, 39–49 (1964).
Galindo, L. A., Puillandre, N., Utge, J., Lozouet, P. & Bouchet, P. The phylogeny and systematics of the Nassariidae revisited (Gastropoda, Buccinoidea). Mol. Phylogenet. Evol. 99, 337–353. https://doi.org/10.1016/j.ympev.2016.03.019 (2016).
Poppe, G. & Goto, Y. European Seashells Vol. 1 (Vera Christa Hemmen, Germany, 1991).
Gofas, S., Moreno, D. & Salas, C. Moluscos Marinos de Andalucía. (Servicio de Publicaciones e Intercambio Científico, Universidad de Málaga., 2011).
WoRMS. http://www.marinespecies.org/aphia.php?p=taxdetails&id=246140, accessed 28 January 2019 (2019).
Pérès, J. M. & Picard, J. Nouveau manuel de bionomie benthique. Recl. Trav. Stn. Mar. Endoume 31, 5–137 (1964).
Mars, P. Recherches sur quelques étangs du littoral méditerranéen français et leurs faunes malacologiques. Vie et milieu supp. 20, 359 (1966).
Zaouali, J. Influence des facteurs thermiques et halins sur la faune malacologique de quelques lagunes tunisiennes (lac lchkeul, lac de Bizerte, lac de Tunis, mer de Bou Grara. Rapp. Comm. Int. Mer Medit. 23, 99–101 (1975).
UNEP/MAP-RAC/SPA. Handbook for Interpreting Types of Marine Habitat for the Selection of Sites to be Included in the National Inventories of Natural Sites of Conservation Interest (Bellan-Santini D, Bellan G, Ghazi Bitar G, Harmelin J-G, Pergent ) 217 (2007).
Russo, P. Lagoon malacofauna: results of malacological research in the Venice Lagoon. Boll. Malacol. 53, 49–62 (2017).
Nobre, A. Moluscos Marinhos de Portugal (Imprensa Portuguesa, Porto, 1931).
Grossu, A. V. Gastropoda Prosobranchia şi Opisthobranchia. Fauna Republicii Populare Române. Mollusca, Bucureşti, 3, fasc. 2, p 220. (1956).
Parenzan, P. Carta d’identità delle conchiglie del Mediterraneo. Volume Primo. Gasteropodi. Bios Taras, Taranto, 283 (1970).
Sauriau, P. G. Spread of cyclope-neritea (mollusca, gastropoda) along the north-eastern Atlantic coasts in relation to oyster culture and to climatic fluctuations. Mar. Biol. 109, 299–309. https://doi.org/10.1007/bf01319398 (1991).
Anistratenko, V., Khaliman, I. & Anistratenko, O. The Molluscs of the Sea of Azov, Naukova Dumka, p 186. ISBN: 978-966-00-1112-0. (2011).
Revkov, N. et al. in BSC, State of the Environment of the Black Sea (20012006/7) 243–290. (Black Sea Commission Publications 2008-3, 2008).
Gili, C. & Martinell, J. Phylogeny, speciation and species turnover. The case of the Mediterranean gastropods of genus Cyclope Risso, 1826. Lethaia 33, 236–250. https://doi.org/10.1080/00241160025100080 (2000).
Sabelli, B. & Taviani, M. In The Mediterranean Sea: Its History and Present Challenges (eds Goffredo, S. & Dubinsky, Z.) 285–306 (Springer, Dordrecht, 2014).
Borsa, P. et al. Infraspecific zoogeography of the Mediterranean: population genetic analysis on sixteen atlanto-mediterranean species (fishes and invertebrates). Vie Milieu 47, 295–305 (1997).
Bremer, J. R. A., Vinas, J., Mejuto, J., Ely, B. & Pla, C. Comparative phylogeography of Atlantic bluefin tuna and swordfish: the combined effects of vicariance, secondary contact, introgression, and population expansion on the regional phylogenies of two highly migratory pelagic fishes. Mol. Phylogenet. Evol. 36, 169–187. https://doi.org/10.1016/j.ympev.2004.12.011 (2005).
Patarnello, T., Volckaert, F. & Castilho, R. Pillars of Hercules: is the Atlantic-Mediterranean transition a phylogeographical break?. Mol. Ecol. 16, 4426–4444. https://doi.org/10.1111/j.1365-294X.2007.03477.x (2007).
Maggs, C. A. et al. Evaluating signatures of glacial refugia for north Atlantic benthic marine taxa. Ecology 89, S108–S122. https://doi.org/10.1890/08-0257.1 (2008).
Rolán, E. D. Especies más de moluscos mediterráneos introducidos en la bahía de O Grove. Thalassas 10, 135 (1992).
Bachelet, G., Cazaux, C., Gantès, H. & Labourg, P. Contribution à l’étude de la faune marine de la région d’Arcachon. Bull. Cent. Etudes Rech. Sci. Biarritz IX, 45–64 (1980).
Bachelet, G. et al. Invasion of the eastern Bay of Biscay by the nassariid gastropod Cyclope neritea: origin and effects on resident fauna. Mar. Ecol. Prog. Ser. 276, 147–159. https://doi.org/10.3354/meps276147 (2004).
Simon-Bouhet, B., Garcia-Meunier, P. & Viard, F. Multiple introductions promote range expansion of the mollusc Cyclope neritea (Nassariidae) in France: evidence from mitochondrial sequence data. Mol. Ecol. 15, 1699–1711. https://doi.org/10.1111/j.1365-294X.2006.02881.x (2006).
Couceiro, L., Miguez, A., Ruiz, J. M. & Barreiro, R. Introduced status of Cyclope neritea (Gastropoda, Nassariidae) in the NW Iberian Peninsula confirmed by mitochondrial sequence data. Mar. Ecol. Prog. Ser. 354, 141–146. https://doi.org/10.3354/meps07257 (2008).
Simon-Bouhet, B., Daguin, C., Garcia-Meunier, P. & Viard, F. Polymorphic microsatellites for the study of newly established populations of the gastropod Cyclope neritea. Mol. Ecol. Notes 5, 121–123. https://doi.org/10.1111/j.1471-8286.2005.00857.x (2005).
Aissaoui, C., Galindo, L. A., Puillandre, N. & Bouchet, P. The nassariids from the Gulf of Gabes revisited (Neogastropoda, Nassariidae). Mar. Biol. Res. 13, 370–389. https://doi.org/10.1080/17451000.2016.1273528 (2017).
Knowlton, N. & Jackson, J. Inbreeding and outbreeding in marine invertebrates. In The Natural History of Inbreeding and Outbreeding: Theoretical and Empirical Perspectives (ed. Thornhill, N. W.) 200–249 (University of Chicago Press, Chicago, 1993).
Cahill, A. E. & Levinton, J. S. Genetic differentiation and reduced genetic diversity at the northern range edge of two species with different dispersal modes. Mol. Ecol. 25, 515–526. https://doi.org/10.1111/mec.13497 (2016).
Cahill, A. E. & Viard, F. Genetic structure in native and non-native populations of the direct-develo** gastropod Crepidula convexa. Mar. Biol. 161, 2433–2443. https://doi.org/10.1007/s00227-014-2519-2 (2014).
Boissin, E. et al. Contemporary genetic structure and postglacial demographic history of the black scorpionfish, Scorpaena porcus, in the Mediterranean and the Black Seas. Mol. Ecol. 25, 2195–2209. https://doi.org/10.1111/mec.13616 (2016).
Simon-Bouhet, B. Expansion d’aire et processus d’introductions biologiques en milieu marin: le cas de Cyclope neritea (Nassariidae) sur les côtes françaises. Thèse de Doctorat, Université de La Rochelle, France, p. 248 (2006).
Couceiro, L., Lopez, L., Ruiz, J. M. & Barreiro, R. Population structure and range expansion: the case of the invasive gastropod Cyclope neritea in northwest Iberian Peninsula. Integr. Zool. 7, 286–298. https://doi.org/10.1111/j.1749-4877.2012.00305.x (2012).
Spalding, M. D. et al. Marine ecoregions of the world: a bioregionalization of coastal and shelf areas. Bioscience 57, 573–583. https://doi.org/10.1641/b570707 (2007).
Boissin, E., Hoareau, T. B. & Berrebi, P. Effects of current and historic habitat fragmentation on the genetic structure of the sand goby Pomatoschistus minutus (Osteichthys, Gobiidae). Biol. J. Linn. Soc. 102, 175–198. https://doi.org/10.1111/j.1095-8312.2010.01565.x (2011).
Taviani, M. The Mediterranean benthos from Late Miocene up to Present: ten million years of dramatic climatic and geological vicissitudes. Biol. Mar. Mediterr. 9, 445–463 (2002).
Marino, I. A. M., Pujolar, J. M. & Zane, L. Reconciling deep calibration and demographic history: Bayesian inference of post glacial colonization patterns in Carcinus aestuarii (Nardo, 1847) and C. maenas (Linnaeus, 1758). PLoS ONE 6, 10. https://doi.org/10.1371/journal.pone.0028567 (2011).
Grant, W. S., Liu, M., Gao, T. X. & Yanagimoto, T. Limits of Bayesian skyline plot analysis of mtDNA sequences to infer historical demographies in Pacific herring (and other species). Mol. Phylogenet. Evol. 65, 203–212. https://doi.org/10.1016/j.ympev.2012.06.006 (2012).
Silva, G., Horne, J. B. & Castilho, R. Anchovies go north and west without losing diversity: post-glacial range expansions in a small pelagic fish. J. Biogeogr. 41, 1171–1182. https://doi.org/10.1111/jbi.12275 (2014).
Albaina, N., Olsen, J. L., Couceiro, L., Ruiz, J. M. & Barreiro, R. Recent history of the European Nassarius nitidus (Gastropoda): phylogeographic evidence of glacial refugia and colonization pathways. Mar. Biol. 159, 1871–1884. https://doi.org/10.1007/s00227-012-1975-9 (2012).
Krijgsman, W. et al. Quaternary time scales for the Pontocaspian domain: interbasinal connectivity and faunal evolution. Earth Sci. Rev. 188, 1–40. https://doi.org/10.1016/j.earscirev.2018.10.013 (2018).
Buyukmeric, Y. Postglacial floodings of the Marmara Sea: molluscs and sediments tell the story. Geomar. Lett. 36, 307–321. https://doi.org/10.1007/s00367-016-0446-6 (2016).
Semikolennykh, D., Ignatov, E., Yanina T. & Arslanov, K. Malacofauna of the Kerch Strait during the Late Pleistocene-Holocene: paleogeographical analysis. In: IGCP 610 Fourth Plenary Conference and Field Trip, Tbilisi, Georgia, 2–9 October 2016, 149–152 (2016).
Samadi, S., Lambourdiere, J., Hebert, P. & Boisselier-Dubayle, M. C. Polymorphic microsatellites for the study of adults, egg-masses and hatchlings of five Cerithium species (Gastropoda) from the Mediterranean sea. Mol. Ecol. Notes 1, 44–46. https://doi.org/10.1046/j.1471-8278.2000.00019.x (2001).
Ribeiro, P. A., Branco, M., Hawkins, S. J. & Santos, A. M. Recent changes in the distribution of a marine gastropod, Patella rustica, across the Iberian Atlantic coast did not result in diminished genetic diversity or increased connectivity. J. Biogeogr. 37, 1782–1796. https://doi.org/10.1111/j.1365-2699.2010.02330.x (2010).
Cossu, P. et al. Surviving at the edge of a fragmented range: patterns of genetic diversity in isolated populations of the endangered giant Mediterranean limpet (Patella ferruginea). Mar. Biol. 164, 18. https://doi.org/10.1007/s00227-017-3080-6 (2017).
Dupont, L., Bernas, D. & Viard, F. Sex and genetic structure across age groups in populations of the European marine invasive mollusc, Crepidula fornicata L. (Gastropoda). Biol. J. Linn. Soc. 90, 365–374. https://doi.org/10.1111/j.1095-8312.2007.00731.x (2007).
Paterno, M. et al. A genome-wide approach to the phylogeography of the mussel Mytilus galloprovincialis in the Adriatic and the Black Seas. Front. Mar. Sci. 6, 16. https://doi.org/10.3389/fmars.2019.00566 (2019).
Hare, M. P., Karl, S. A. & Avise, J. C. Anonymous nuclear DNA markers in the American oyster and their implications for the heterozygote deficiency phenomenon in marine bivalves. Mol. Biol. Evol. 13, 334–345. https://doi.org/10.1093/oxfordjournals.molbev.a025593 (1996).
Johnson, M. S. & Black, R. The Wahlund effect and the geographical scale of variation in the intertidal limpet Siphonaria sp. Mar. Biol. 79, 295–302. https://doi.org/10.1007/bf00393261 (1984).
Mallet, A. L., Zouros, E., Gartnerkepkay, K. E., Freeman, K. R. & Dickie, L. M. Larval viability and heterozygote deficiency in populations of marine bivalves—evidence from pair matings of mussels. Mar. Biol. 87, 165–172. https://doi.org/10.1007/bf00539424 (1985).
Boissin, E., Hoareau, T. B., Feral, J. P. & Chenuil, A. Extreme selfing rates in the cosmopolitan brittle star species complex Amphipholis squamata: data from progeny-array and heterozygote deficiency. Mar. Ecol. Prog. Ser. 361, 151–159. https://doi.org/10.3354/meps07411 (2008).
Boissin, E., Egea, E., Feral, J. P. & Chenuil, A. Contrasting population genetic structures in Amphipholis squamata, a complex of brooding, self-reproducing sister species sharing life history traits. Mar. Ecol. Prog. Ser. 539, 165–177. https://doi.org/10.3354/meps11480 (2015).
Dudu, A., Georgescu, S. E., Suciu, R., Dinischiotu, A. & Costache, M. Microsatelitte DNA variation in the black sea beluga sturgeon (Huso huso). Rom. Biotech. Lett. 13, 3779–3783 (2008).
Wilson, A. B. & Veraguth, I. E. The impact of Pleistocene glaciation across the range of a widespread European coastal species. Mol. Ecol. 19, 4535–4553. https://doi.org/10.1111/j.1365-294X.2010.04811.x (2010).
Limborg, M. T. et al. Imprints from genetic drift and mutation imply relative divergence times across marine transition zones in a pan-European small pelagic fish (Sprattus sprattus). Heredity 109, 96–107. https://doi.org/10.1038/hdy.2012.18 (2012).
Miralles, L., Juanes, F., Pardinas, A. F. & Garcia-Vazquez, E. Paleoclimate shaped bluefish structure in the northern hemisphere. Fisheries 39, 578–586. https://doi.org/10.1080/03632415.2014.976701 (2014).
Magoulas, A., Castilho, R., Caetano, S., Marcato, S. & Patarnello, T. Mitochondrial DNA reveals a mosaic pattern of phylogeographical structure in Atlantic and Mediterranean populations of anchovy (Engraulis encrasicolus). Mol. Phylogenet. Evol. 39, 734–746. https://doi.org/10.1016/j.ympev.2006.01.016 (2006).
Durand, J. D., Blel, H., Shen, K. N., Koutrakis, E. T. & Guinand, B. Population genetic structure of Mugil cephalus in the Mediterranean and Black Seas: a single mitochondrial clade and many nuclear barriers. Mar. Ecol. Prog. Ser. 474, 243–261. https://doi.org/10.3354/meps10080 (2013).
Pascual, M., Rives, B., Schunter, C. & Macpherson, E. Impact of life history traits on gene flow: a multispecies systematic review across oceanographic barriers in the Mediterranean Sea. PLoS ONE 12, 20. https://doi.org/10.1371/journal.pone.0176419 (2017).
Anderson, E. C. & Dunham, K. K. The influence of family groups on inferences made with the program Structure. Mol. Ecol. Resour. 8, 1219–1229. https://doi.org/10.1111/j.1755-0998.2008.02355.x (2008).
Peterman, W., Brocato, E. R., Semlitsch, R. D. & Eggert, L. S. Reducing bias in population and landscape genetic inferences: the effects of sampling related individuals and multiple life stages. PeerJ 4, 19. https://doi.org/10.7717/peerj.1813 (2016).
Waples, R. S. & Anderson, E. C. Purging putative siblings from population genetic data sets: a cautionary view. Mol. Ecol. 26, 1211–1224. https://doi.org/10.1111/mec.14022 (2017).
Fernandez, R., Lemer, S., McIntyre, E. & Giribet, G. Comparative phylogeography and population genetic structure of three widespread mollusc species in the Mediterranean and near Atlantic. Mar. Ecol. Evol. Perspect. 36, 701–715. https://doi.org/10.1111/maec.12178 (2015).
Selwyn, J. D. et al. Kin-aggregations explain chaotic genetic patchiness, a commonly observed genetic pattern, in a marine fish. PLoS ONE 11, 11. https://doi.org/10.1371/journal.pone.0153381 (2016).
Highsmith, R. C. Floating and algal rafting as potential dispersal mechanisms in brooding invertebrates. Mar. Ecol. Prog. Ser. 25, 169–179. https://doi.org/10.3354/meps025169 (1985).
Thiel, M. & Haye, P. A. In Oceanography and Marine Biology—An Annual Review Vol. 44 (eds Gibson, R. N. et al.) 323–429 (CRC Press-Taylor & Francis Group, Boca Raton, 2006).
Darras, H. & Aron, S. Introgression of mitochondrial DNA among lineages in a hybridogenetic ant. Biol. Lett. 11, 4. https://doi.org/10.1098/rsbl.2014.0971 (2015).
Perea, S., Vukic, J., Sanda, R. & Doadrio, I. Ancient mitochondrial capture as factor promoting mitonuclear discordance in freshwater fishes: a case study in the genus Squalius (Actinopterygii, Cyprinidae) in Greece. PLoS ONE 11, 26. https://doi.org/10.1371/journal.pone.0166292 (2016).
Markova, S., Dufresne, F., Manca, M. & Kotlik, P. Mitochondrial capture misleads about ecological speciation in the Daphnia pulex complex. PLoS ONE 8, 14. https://doi.org/10.1371/journal.pone.0069497 (2013).
Rawson, P. D. & Hilbish, T. J. Asymmetric introgression of mitochondrial DNA among European populations of blue mussels (Mytilus spp.). Evolution 52, 100–108. https://doi.org/10.2307/2410924 (1998).
Azuma, N., Yamazaki, T. & Chiba, S. Mitochondrial and nuclear DNA analysis revealed a cryptic species and genetic introgression in Littorina sitkana (Mollusca, Gastropoda). Genetica 139, 1399–1408. https://doi.org/10.1007/s10709-012-9638-9 (2011).
Rius, M. & Darling, J. A. How important is intraspecific genetic admixture to the success of colonising populations?. Trends Ecol. Evol. 29, 233–242. https://doi.org/10.1016/j.tree.2014.02.003 (2014).
Boissin, E., Hoareau, T. B., Postaire, B., Gravier-Bonnet, N. & Bourmaud, C. A. F. Cryptic diversity, low connectivity and suspected human-mediated dispersal among 17 widespread Indo-Pacific hydroid species of the south-western Indian Ocean. J. Biogeogr. 45, 2104–2117. https://doi.org/10.1111/jbi.13388 (2018).
Boero, F. et al. CoCoNet: towards coast to coast networks of marine protected areas (from the shore to the high and deep sea), coupled with sea-based wind energy potential. Scires-It 6, 1–95 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 (2009).
Bandelt, H. J., Forster, P. & Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036 (1999).
Nylander, J. MrAIC.pl. Program Distributed by the Author (Evolutionary Biology Centre, Uppsala University, Sweden, 2004).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, 6. https://doi.org/10.1371/journal.pcbi.1003537 (2014).
Wilke, T., Schultheiss, R. & Albrecht, C. As time goes by: a simple fool’s guide to molecular clock approaches in invertebrates. Am. Malacol. Bull. 27, 25–45 (2009).
Stelbrink, B., Shirokaya, A. A., Foller, K., Wilke, T. & Albrecht, C. Origin and diversification of Lake Ohrid’s endemic acroloxid limpets: the role of geography and ecology. BMC Evol. Biol. 16, 13. https://doi.org/10.1186/s12862-016-0826-6 (2016).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: software for identifying and correcting genoty** errors in microsatellite data. Mol. Ecol. Notes 4, 535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x (2004).
Panova, M., Makinen, T., Fokin, M., Andre, C. & Johannesson, K. Microsatellite cross-species amplification in the genus Littorina and detection of null alleles in Littorina saxatilis. J. Molluscan Stud. 74, 111–117. https://doi.org/10.1093/mollus/eym052 (2008).
GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations, Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II, Montpellier (France) (1996–2004).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539. https://doi.org/10.1093/bioinformatics/bts460 (2012).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x (2005).
Earl, D. A. & Vonholdt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. https://doi.org/10.1007/s12686-011-9548-7 (2012).
Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145, 1219–1228 (1997).
Jones, O. R. & Wang, J. L. COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resour. 10, 551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x (2010).
Fu, Y. X. & Li, W. H. Statistical tests of neutrality of mutations. Genetics 133, 693–709 (1993).
Ramos-Onsins, S. E. & Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 19, 2092–2100. https://doi.org/10.1093/oxfordjournals.molbev.a004034 (2002).
Acknowledgements
This project was funded by the European FP7 CoCoNet project (Ocean.2011-4, grant agreement #287844) and we are grateful to the whole CoCoNET consortium. We are grateful to the following people for their critical help with logistics and field work ‘Antheus srl (Lecce, Italy)’; S Bevilacqua, G Guarnieri, S Fraschetti and T Terlizzi (University of Salento, Italy); L Angeletti and M Sigovini (ISMAR, Italy); D Shamrey (IBSS, Sevastopol); A Anastasopoulou, MA Pancucci-Papadopoulou and S Reizopoulou (HCMR, Greece) and E Hajdëri (Catholic University ‘Our Lady of Good Counsel’, Tirana). Thank you to J Almany for English corrections. This is ISMAR-CNR scientific contribution n1987. E Boissin was supported by a European Marie Curie postdoctoral fellowship MC-CIG-618480.
Author information
Authors and Affiliations
Contributions
S.P., L.Z., D.M. conceived the project. D.M., L.B., B.T., V.T., M.P., C.K., N.M., E.V., S.B., I.N., G.A., M.T., S.P. performed the sampling. V.N and S.B. performed the laboratory work. E.B. supervised the laboratory work, performed the analyses and drafted the manuscript with input from D.M., M.T., L.Z., S.P. All authors read and improved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boissin, E., Neglia, V., Baksay, S. et al. Chaotic genetic structure and past demographic expansion of the invasive gastropod Tritia neritea in its native range, the Mediterranean Sea. Sci Rep 10, 21624 (2020). https://doi.org/10.1038/s41598-020-77742-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-77742-3
- Springer Nature Limited