
Abstract
The Bethylidae are the most diverse of Hymenoptera chrysidoid families. As external parasitoids, the bethylids have been widely adopted as biocontrol agents to control insect pests worldwide. Thus far, the genomic information of the family Bethylidae has not been reported yet. In this study, we crystallized into a high-quality chromosome-level genome of ant-like bethylid wasps Sclerodermus sp. ‘alternatusi’ (Hymenoptera: Bethylidae) using PacBio sequencing as well as Hi-C technology. The assembled S. alternatusi genome was 162.30 Mb in size with a contig N50 size of 3.83 Mb and scaffold N50 size of 11.10 Mb. Totally, 92.85% assembled sequences anchored to 15 pseudo-chromosomes. A total of 10,204 protein-coding genes were annotated, and 23.01 Mb repetitive sequences occupying 14.17% of genome were pinpointed. The BUSCO results showed that 97.9% of the complete core Insecta genes were identified in the genome, while 97.1% in the gene sets. The high-quality genome of S. alternatusi will not only provide valuable genomic information, but also show insights into parasitoid wasp evolution and bio-control application in future studies.
Similar content being viewed by others

Background & Summary
Parasitic Hymenoptera is hyperdiverse in the insect lineages and comprises a prominent plurality of venomous species1. This group have evolved diversified parasitic strategies to manipulate their hosts, such as producing venom2, polydnavirus3, teratocytes4, ovarian proteins5, and larval secretions, leading to host killed or paralyzed permanently6. Parasitoid wasps provide a sustainable approach in biocontrol of insect pests, thus conferring enormous economic and ecological benefits to global agriculture and forestry7,8. For instance, an obligate aphid parasitoid Aphidius gifuensis (Hymenoptera: Braconidae) was applied to control the green peach aphid Myzus persicae, one of the most economically important aphid crop pests worldwide9,10. The pupal endoparasitoid Trichopria drosophilae (Hymenoptera: Diapriidae) was used to control the spotted wing drosophila Drosophila suzukii, a fruit fly that causes massive economic damage to a variety of summer fruit in United States11. Recently, parasitoid wasps are instantly develo** as a promising model to gain insight into genome size evolution and parasite-host coevolution12,13.
The Bethylidae are the most diverse of Hymenoptera chrysidoid families, with more than 3,000 external parasitoids of Lepidopteran and Coleopteran larvae14. The bethylids have been widely adopted as biocontrol agents to control insect pests worldwide. The ant-like bethylid wasps Sclerodermus sp. ‘alternatusi’ and S. guani (Hymenoptera: Bethylidae) are ectoparasitoid wasps indigenous to China15. The adults S. alternatusi are brown-colored insects with stout legs, morphologically resembling S. guani. The head is distinctly prognathous and 13 antennomers in both sexes. The eye length of males is a little more than half the length of the head and gibbose, but the eyes of females are reduced. Females tend to be wingless (Fig. 1a,b), whereas males are mostly winged with strongly-reduced veins (Fig. 1c,d). The metasoma has seven or eight abdominal segments externally visible. Both species generally parasitize the larva of wood-boring insects such as Monochamus alternatus, which vectors pinewood nematode Bursaphelenchus xylophilus, the causal agent of pine wilt disease16,17. The wood-boring insects are difficult to control, due to its hidden early life, conceal in their habits (trunk, wood, or seed) and long emergence period of the adults18. S. alternatusi possesses the ability of invading the chamber structures and detecting host larvae or pupae. Females secrete venom from venom reservoir and inject venom subsequently to permanently paralyze the hosts prior to feeding and oviposition. After that, S. alternatusi larvae (Fig. 2a) absorb nutrition from the hosts until adult emergence19,20. During this process, females protect the eggs and larvae by moving their location upon the host externally. Because of its high parasitism rate and easy artificial rearing (Fig. 2b), S. alternatusi has been mass bred on a commercial scale21.

Morphology of S. alternatusi adults. (a) Dorsal view of a wingless female. (b) Ventral view of a wingless female. (c) Dorsal view of a winged male. (d) Ventral view of a winged male.

Life cycle of S. alternatusi and workflow used in the genome sequencing and assembly. (a) A parasitized larva of the longhorn beetle Thyestilla gebleri. Both S. alternatusi female and larvae are shown. (b) Laboratory rearing of S. alternatusi using a substitute host T. molitor pupae. The winged ratio of males and females is 90% and 50%, respectively. (c) The workflow overview of S. alternatusi chromosome-level genome assembly.
High-quality assembled genomes contribute to the molecular mechanisms behind parasitic biology. Generally, Hymenopteran parasitoids possess a particular haplodiploid sex determination system, where males are haploid and females are diploid. As such, Hymenopteran males were commonly used for genome sequencing to simplify the genome assembly and annotation due to the lack of heterozygosity22. Thus far, more than 100 parasitoid wasps with their genomes sequenced and assembled have been reported, mainly from the family Micryoidea, Ichneumonoidea, Cynipoidea, Cyanoidea and Orussoidea23. Among these, 17 species were sequenced using high-throughput chromosome conformation capture (Hi-C) technology (Table 1). However, the genomic information of the family Bethylidae has not yet been reported.
To gain insights into the evolution of S. alternatusi and the complex relationship between the parasitoid and its hosts, we herein developed a high-quality chromosome-level assembly of the S. alternatusi reference genome. We integrated PacBio sequencing and Hi-C technology (Fig. 2c) for genome assembly. The S. alternatusi genome size is 162.3 Mb with a contig N50 size of 3.83 Mb and scaffold N50 size of 11.1 Mb (Table 2). Notably, 23,014,663 bp repetitive sequences were identified, occupying 14.17% of the S. alternatusi genome size (Table 3). Applying Hi-C scaffolding, we assigned 92.85% bases to 15 pseudo-chromosomes, which was further corroborated by karyoty** analysis (2n = 30). In addition, multiple transcriptome data and homologue protein sequences assisted us in annotating 10,204 protein-coding genes (Table 2). For functional annotation of protein-coding genes, we aligned gene sequences to NR, NT, SwissPro, KOG, eggNOG and InterPro databases, in which 10,027, 6,300, 7,515, 5,997, 9,488 and 9,190 genes were successfully mapped (Table 4), respectively.
Methods
Insect
The parasitoid wasp S. alternatusi colony was maintained in our laboratory, Zhejiang University, Hangzhou, China. Wasps were reared on a substitute host, the yellow mealworm beetle Tenebrio molitor (Fig. 2b), and maintained at 26 °C and 70% relative humidity in the 5 mL finger-shaped tubes with cotton plugged.
Genome survey
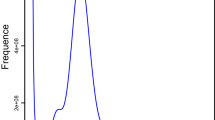
Qualified genomic DNA (gDNA) was extracted from 300 female adults. A DNA library was constructed using TruSeq Nano DNA library kit (Illumina, USA), with an average insert size of 350 bp. The library was sequenced using Illumina NovaSeq 6000 platform from Annoroad gene technology Co., Ltd. (Bei**g, China). To obtain clean reads, raw data were filtered by removing low quality, short reads, cut adapters, and polyG. In total, 52.64 Gb clean reads were maintained for subsequent survey analysis. The genome size, heterozygosity, and repeat content of S. alternatusi were estimated by Genomescope v2.024 (https://github.com/tbenavi1/genomescope2.0), and the results were visualized by K-mer (k = 17, produced by Jellyfish v2.2.10) frequency distribution map25 (Fig. 3a). The estimated genome size of S. alternatusi was 180.86 Mb, with the estimated genome repeat length of 51.56 Mb and the estimated heterozygosity of 0.157%.

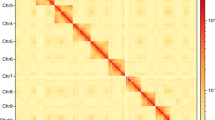
Characteristics of the S. alternatusi genome. (a) S. alternatusi genome size estimation by K-mer distribution (K = 17). The x-axis represents the K-mer depth and the y-axis is the corresponding frequency. (b) Circos plot of the S. alternatusi genome assembly. Tracks from inner to outer represent the following: (1) 15 pseudo-chromosomes at the Mb scale; (2) GC content; and (3) genes density. (c) Hi-C heatmap of S. alternatusi. The scale bar shows all interaction frequency of 15 chromosomes. (d) Karyogram of S. alternatusi: 2n = 30.
PacBio library construction and de novo assembly
gDNA was extracted from 1,000 female adults using FineOut animal tissue DNA kit (Genfine, China). The DNA integrity was assessed by Agilent 4200 bioanalyser (Agilent Technologies, China). Subsequently, Megaruptor 3 (Diagenode, USA) was applied to shear the gDNA, followed by purification using AMPure PB magnetic beads (Pacific Biosciences, USA). Each SMRTbell library was constructed using the PacBio SMRTbell template prep kit 2.0 (Pacific Biosciences, USA). The BluePippin system was utilized for DNA size selection. The genome of the S. alternatusi was sequence on PacBio Sequel II platform, and the output data was visualized using SMRTlink v11.0 (PacBio, USA). A total of 31.77 Gb PacBio CCS (HiFi) reads (after error correction from 546.19 Gb raw data) were obtained with an average length of 16.86 kb and an N50 length of 17.73 kb, which was further used for de novo assembly. The assembler software Wtdbg2 v2.526 (https://github.com/ruanjue/wtdbg2-xsq-g180m) delivered initial contigs of high-quality assembly from S. alternatusi HiFi reads. The genome size of the first assembly was 180,768,149 bp with a contig N50 of 3,562,991 bp and an Insecta BUSCO completeness of 97.8% (95.1% single-copy and 2.7% duplicated genes). Purge Haplotigs v1.1.127 (https://bitbucket.org/mroachawri/purge_haplotigs/src/-a60) combined with HaploMerger2 v3.628 with default parameters were employed to remove heterozygosity from the first assembly. The NCBI non-redundant nucleotide database (NT) was used to identify and eliminate possible contaminating sequences. The pseudo-haplotype assembly was 162,151,154 bp with a contig N50 of 4,324,342 bp and an Insecta BUSCO completeness of 97.8% (95.8% single-copy and 2.0% duplicated genes).
Hi-C library construction and sequencing
Chromosome contact information was revealed from Hi-C data. A total of 70 S. alternatusi larvae were collected for preparing Hi-C libraries according to standard protocols29. The samples were crosslinked with 2% formaldehyde solution at room temperature for 10 min and then added with 2.5 M-glycine solution priority to quality control. Hi-C libraries were constructed and sequenced using Illumina Novaseq 6000 platform. Hi-C data were used to anchor the contigs to chromosomes as well as orienting the scaffolds into super scaffolds. We used SAMtools (https://github.com/samtools) and Chromap30 with default parameters (https://github.com/haowenz/chromap) to align Hi-C clean reads with the assembled S. alternatusi genome. Then YahS31 with default parameters (https://github.com/c-zhou/yahs) was used to construct chromosome-scale scaffolds. The contact frequency matrix results were obtained with Juicer tools v1.19.02 (https://github.com/aidenlab/juicer), followed by visualization of contigs and scaffolds using JuiceBox v1.11.0832 with default parameters (https://github.com/aidenlab/Juicebox). Ultimately, the size of the S. alternatusi genome was 162.30 Mb with a contig N50 of 3.83 Mb (Table 2), and the contigs were anchored to 15 pseudo-chromosomes (Fig. 3b,c, and Table 5).
Cytogenetic karyotype analysis
Heads were dissected from 4–6 d larvae (n = 20) and then mixed with 1 mL of 0.07–0.08 mg/mL colchicine at 25 °C for 3 h. The tissues were treated with 1% hypotonic sodium citrate solution for 1 h, followed by immobilizing with a fixative solution (methanol: acetic acid, 3:1) at 4 °C for 1 h. The tissues were softened in 60% acetic acid for 30 min and then fixed in the fixative solution again for 10 min. Subsequently, samples were ground in the fixative solution using disposable tissue grinding pestles. The cell suspension was dropped onto a pre-chilled glass slide. After being air-dried, the cells were stained with 5 µg/mL DAPI for 5 min and rinsed with running water. Chromosomes (2n = 30) were observed using an Olympus FV3000 microscope with 60 × magnification (Fig. 3d).
Transcriptome sequencing
For assisting gene annotation, we prepared transcriptomes of S. alternatusi from three developmental stages including eggs, pupae (4d, 8d,and 11d), adult females, and adult males. In addition, four representative tissues including head, fat body, ovary and venom glands were dissected from females for transcriptome sequencing. RNAs were extracted using RNAiso Plus (Takara, China) according to the manufacturer’s protocol. An RNA library was constructed using NEBNext ultra RNA library prep kit (NEB, USA) following the manufacturer’s recommendations. RNA sequencing was performed on the Illumina Novaseq. 6000 platform. Full-length transcripts were assembled by Trinity v2.15.133 with default parameters (https://github.com/trinityrnaseq).
Gene prediction and annotation
A genome-wide annotation tool (GWAT) GETA v2.4.1 with default parameters (https://github.com/chenlianfu/geta) was employed to perform gene prediction and annotation. RepeatMasker v4.1.2 with default parameters (http://repeatmasker.org/RepeatMasker) was applied to identify the repetitive sequences, encompassing interspersed repeats and transposable elements (TEs). Then RepeatModeler v2.0.3 with default parameters (http://www.repeatmasker.org/RepeatModeler) and Repbase library34 (https://www.girinst.org/repbase) assisted to cluster the repeats by building a de novo repeat library. A total of 23,014,663 bp repetitive sequences were obtained, accounting for 14.17% genome size. Four classes of TEs, including long terminal repeats (LTRs), long interspersed nuclear elements (LINEs), DNA elements (DNAs) and short interspersed nuclear elements (SINEs), comprised 1.42%, 0.54%, 3.10% and 0.01% of the S. alternatusi genome, respectively (Table 3).
After masking the repeat sequences, the S. alternatusi genome was annotated by integrating evidence including ab initio gene predictions, transcripts and protein homologues. Augustus v3.4.035 with default parameters (https://github.com/Gaius-Augustus/Augustus) was applied for ab initio gene predictions. Then PASA v2.4.136 with default parameters (https://github.com/PASApipeline) was employed to align the transcriptome data to the genome, followed by homology-based prediction using GeneWise v2.4.137 with default parameters (https://www.ebi.ac.uk/Tools/psa/genewise). EVidenceModeler v1.1.138 with default parameters (https://github.com/EVidenceModeler/EVidenceModeler) was employed to integrate the output from the above approaches to generate a combined annotation model. Furthermore, functional annotation of the predicted protein-coding gene was carried out by searching against the NCBI non-redundant databases (NR) (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz), the nucleotide sequence database (NT) (https://www.ncbi.nlm.nih.gov/nucleotide), SwissProt39, eukaryotic orthologous groups (KOG)40 as well as InterPro41. The genes were also mapped to eggNOG database42. Ultimately, a total of 10,204 protein-coding genes in the genome were successfully annotated (Table 4).
Data Records
All data generated during this study including genome assembly, transcriptome assembly and raw sequencing data were submitted to NCBI. The raw sequencing data have been deposited at NCBI Sequence Read Archive (SRA) under BioProject accession PRJNA1087141 with the accession number SRP49506643. This Whole Genome Shotgun project has been deposited at GenBank under the accession JBBEEM000000000. The version described in this paper is version JBBEEM01000000044. The whole sequencing dataset and genome assembly reported in this paper have been also deposited in the Genome Sequence Archive (GSA) at the National Genomics Data Center (NGDC)/China National Center for Bioinformation (CNCB) under accession number CRA01252645. The genome annotation has been deposited in the Genome Warehouse46 in National Genomics Data Center47, Bei**g Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation, under accession number GWHEQBB00000000.
Technical Validation
The Hi-C intra-chromosomal contact map with high alignment ratio (92.85%), indicated valid interaction information of 15 pseudo-chromosomes such as homologous contact pattern of chromosomes and translocated regions. The chromosome-level genome assembly quality of S. alternatusi was evaluated by performing BUSCO v5.4.648 with default parameters, which presented that 97.9% of BUSCO genes (insecta_db10) were successfully identified in the genome assembly, encompassing complete and single-copy (96%), complete and duplicated (1.9%), fragmented (0.1%) and missing (2%) categories. The gene annotation result was assessed using BUSCO, indicating 1,327 (97.1%) genes were functional annotated. In summary, we provide a high-quality genome with high level of completeness and accuracy.
Code availability
The command and pipelines used for data analyses in this study were executed according to corresponding protocols of bioinformatics software. No custom programming or coding was used. The version and parameters have been mentioned in Methods.
References
Dashevsky, D. et al. Functional and Proteomic Insights into Aculeata Venoms. Toxins (Basel) 15, 224–249 (2023).
Parkinson, N. M. et al. Towards a comprehensive view of the primary structure of venom proteins from the parasitoid wasp Pimpla hypochondriaca. Insect Biochem. Mol. Biol. 34, 565–571 (2004).
Belle, E. et al. Visualization of Polydnavirus Sequences in a Parasitoid Wasp Chromosome. J. Virol. 76, 5793–5796 (2002).
Dahlman, D. L. et al. A teratocyte gene from a parasitic wasp that is associated with inhibition of insect growth and development inhibits host protein synthesis. Insect Mol. Biol. 12, 527–534 (2003).
Luckhart, S. & Webb, B. A. Interaction of a wasp ovarian protein and polydnavirus in host immune suppression. Dev. Comp. Immunol. 20, 1–21 (1996).
Pennacchio, F. & Strand, M. R. Evolution of developmental strategies in parasitic hymenoptera. Annu. Rev. Entomol. 51, 233–258 (2006).
Wang, Z., Liu, Y., Shi, M., Huang, J. & Chen, X. Parasitoid wasps as effective biological control agents. J. Integr. Agric. 18, 705–715 (2019).
Polaszek, A. & Vilhemsen, L. Biodiversity of hymenopteran parasitoids. Curr. Opin. Insect Sci. 56, 1–7 (2023).
Li, B. et al. Chromosome-level genome assembly of the aphid parasitoid Aphidius gifuensis using Oxford Nanopore sequencing and Hi-C technology. Mol. Eco.l Resour. 21, 941–954 (2021).
Singh, K. S. et al. Global patterns in genomic diversity underpinning the evolution of insecticide resistance in the aphid crop pest Myzus persicae. Commun. Biol. 4, 847 (2021).
Häussling, B. J. M., Lienenlüke, J. & Stökl, J. The preference of Trichopria drosophilae for pupae of Drosophila suzukii is independent of host size. Sci. Rep. 11, 1–10 (2021).
Ye, X. et al. Genomic signatures associated with maintenance of genome stability and venom turnover in two parasitoid wasps. Nat. Commun. 13, 6417 (2022).
Beckage, N. E. & Gelman, D. B. Wasp Parasitoid Disruption of Host Development: Implications for New Biologically Based Strategies for Insect Control. Annu. Rev. Entomol. 49, 299–330 (2004).
Brazidec, M. & Perrichot, V. Two new chrysidoid wasps (Hymenoptera: Bethylidae, Chrysididae) from mid-Miocene Zhangpu amber. Palaeoworld 32, 699–708 (2023).
Tang, Y. et al. Parasitism Ability and Offspring Development of Sclerodermus alternatusi Yang (Hymenoptera: Bethylidae) in Different Female Oviposition Times. Chin. J. Biol. Control 39, 499–506 (2023).
Wang, S. Y., Hackney, P. J. & Zhang, D. Hydrocarbons catalysed by TmCYP4G122 and TmCYP4G123 in Tenebrio molitor modulate the olfactory response of the parasitoid Scleroderma guani. Insect Mol. Biol. 28, 637–648 (2019).
Li, Z. et al. The ectoparasitoid Scleroderma guani (Hymenoptera: Bethylidae) uses innate and learned chemical cues to locate its host, larvae of the pine sawyer Monochamus alternatus (Coleoptera: Cerambycidae). Fla. Entomol. 98, 1182–1187 (2015).
Luo, C. W. & Chen, Y. Phototactic Behavior of Scleroderma guani (Hymenoptera: Bethylidae) - Parasitoid of Pissodes punctatus (Coleoptera: Curculionidae). J. Insect Behav. 29, 605–614 (2016).
Li, L., Wei, W., Liu, Z. & Sun, J. Host adaptation of a gregarious parasitoid Sclerodermus harmandi in artificial rearing. BioControl 55, 465–472 (2010).
Lauzière, I., Brodeur, J. & Pérez-Lachaud, G. Host stage selection and suitability in Cephalonomia stephanoderis betrem (hymenoptera: Bethylidae), a parasitoid of the coffee berry borer. Biol. Control 21, 128–133 (2001).
Li, L. F. et al. Parasitism and venom of ectoparasitoid Scleroderma guani impairs host cellular immunity. Arch. Insect Biochem. Physiol. 98, 1–13 (2018).
Yahav, T. & Privman, E. A comparative analysis of methods for de novo assembly of hymenopteran genomes using either haploid or diploid samples. Sci. Rep. 9, 6480 (2019).
Mei, Y. et al. InsectBase 2.0: A comprehensive gene resource for insects. Nucleic Acids Res. 50, 1040–1045 (2022).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Marçais, G., Kingsford, C. & Bateman, A. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, 460 (2018).
Huang, S., Kang, M. & Xu, A. HaploMerger2: Rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579 (2017).
Nagano, T. et al. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol. 16, 175 (2015).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 12, 6566 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2022).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst 6, 256–258 (2018).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8, 1494–1512 (2013).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, (2015).
Stanke, M. et al. AUGUSTUS: A b initio prediction of alternative transcripts. Nucleic Acids Res 34, (2006).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666 (2003).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, (2008).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 45–48 (2000).
Tatusov, R. L. et al. The COG database: An updated vesion includes eukaryotes. BMC Bioinformatics 4, 1–14 (2003).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res 45, 190–199 (2017).
Huerta-Cepas, J. et al. EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, 309–314 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP495066 (2024).
Wan, Y. et al. Sclerodermus sp. ‘alternatusi’ isolate ZJU-2024a, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBBEEM000000000 (2024).
NGDC/CNCB Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA012526 (2023).
Chen, M. et al. Genome Warehouse: A Public Repository Housing Genome-scale Data. Genomics Proteomics Bioinformatics 19, 584–589 (2021).
Xue, Y. et al. Database resources of the national genomics data center, china national center for bioinformation in 2021. Nucleic Acids Res. 49, 18–28 (2021).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 1, e323 (2021).
Ye, X. et al. A chromosome-level genome assembly of the parasitoid wasp Pteromalus puparum. Mol. Ecol. Resour. 20, 1384–1402 (2020).
Wittmeyer, K. T., Oppenheim, S. J. & Hopper, K. R. Assemblies of the genomes of parasitic wasps using meta-assembly and scaffolding with genetic linkage. G3 (Bethesda) 12, jkad386 (2022).
Ye, X. et al. Genome of the parasitoid wasp Cotesia chilonis sheds light on amino acid resource exploitation. BMC Biol. 20, 1–17 (2022).
Gauthier, J. et al. Chromosomal scale assembly of parasitic wasp genome reveals symbiotic virus colonization. Commun. Biol. 4, 104 (2021).
Pinto, B. J. et al. A Chromosome-Level Genome Assembly of the Parasitoid Wasp, Cotesia glomerata (Hymenoptera: Braconidae). J. Hered 112, 558–564 (2021).
Mao, M. et al. A chromosome scale assembly of the parasitoid wasp Venturia canescens provides insight into the process of virus domestication. G3 (Bethesda) 13, jkad137 (2023).
**ao, S. et al. Genome assembly of the ectoparasitoid wasp Theocolax elegans. Sci. Data 10, 159 (2023).
Kuang, J. G. et al. Chromosome-level de novo genome assembly of two conifer-parasitic wasps, Megastigmus duclouxiana and Megastigmus sabinae, reveals genomic imprints of adaptation to hosts. Mol. Ecol. Resour. 23, 1142–1154 (2023).
Shu, X. et al. Chromosome-level genome assembly of Microplitis manilae Ashmead, 1904 (Hymenoptera: Braconidae). Sci. Data 10, 226 (2023).
Dalla Benetta, E. et al. Genome elimination mediated by gene expression from a selfish chromosome. Sci. Adv. 6, eaaz9808 (2020).
Inwood, S. N. et al. Chromosome-level genome assemblies of two parasitoid biocontrol wasps reveal the parthenogenesis mechanism and an associated novel virus. BMC Genom. 24, 440 (2023).
Acknowledgements
We thank Dr. Yang Mei (Zhejiang university) for hel** with data analysis. This research was supported by key projects of Natural Science Foundation of Zhejiang Province (LZ21C140002), National Natural Science Foundation of China (32102186 and 32272519) and Key Research and Development Program of Zhejiang Province (2023C02033).
Author information
Authors and Affiliations
Contributions
H.J.X. convinced and designed the study. Y.W., H.J.W. and J.P.Y. reared the S. alternatusi, Y.W. collected the samples. Y.X.Y., Y.W. and J.L.Z. performed bioinformatics analysis. Y.W. wrote the draft manuscript. H.J.X., Z.C.S. and Y.X.Y. modified the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wan, Y., Wu, HJ., Yang, JP. et al. Chromosome-level genome assembly of the bethylid ectoparasitoid wasp Sclerodermus sp. ‘alternatusi’. Sci Data 11, 438 (2024). https://doi.org/10.1038/s41597-024-03278-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03278-0
- Springer Nature Limited