

Abstract
It is estimated that 10%–30% of disease-associated genetic variants affect splicing. Splicing variants may generate deleteriously altered gene product and are potential therapeutic targets. However, systematic diagnosis or prediction of splicing variants is yet to be established, especially for the near-exon intronic splice region. The major challenge lies in the redundant and ill-defined branch sites and other splicing motifs therein. Here, we carried out unbiased massively parallel splicing assays on 5,307 disease-associated variants that overlapped with branch sites and collected 5,884 variants across the 5′ splice region. We found that strong splice sites and exonic features preserve splicing from intronic sequence variation. Whereas the splice-altering mechanism of the 3′ intronic variants is complex, that of the 5′ is mainly splice-site destruction. Statistical learning combined with these molecular features allows precise prediction of altered splicing from an intronic variant. This statistical model provides the identity and ranking of biological features that determine splicing, which serves as transferable knowledge and out-performs the benchmarking predictive tool. Moreover, we demonstrated that intronic splicing variants may associate with disease risks in the human population. Our study elucidates the mechanism of splicing response of intronic variants, which classify disease-associated splicing variants for the promise of precision medicine.







Similar content being viewed by others


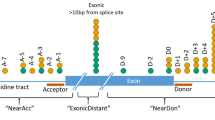
Data availability
RNA Sequencing datasets generated during this study are available at the NCBI GEO: GSE179892. Other databases used in the study: UCSC PhyloP: https://genome.ucsc.edu/cgi-bin/hgTracks?hgsid=1351580935_14MOQtNDW7V78RaXEDp3Yy4m4PTb&c=chr2&hgTracksConfigPage=configure&hgtgroup_compGeno_close=0#compGenoGroup ATtRACT: https://attract.cnic.es/download; Ensembl: https://asia.ensembl.org/info/data/ftp/index.html.Source data are provided with this paper. Further information and requests for resources should be directed to and will be fulfilled by the corresponding author.
Code availability
Custom codes and the training features used in the study are available at https://github.com/chienlinglin/modeling-intron-variants/.
References
Baralle, F. E. & Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18, 437–451 (2017).
Wilkinson, M. E., Charenton, C. & Nagai, K. RNA splicing by the spliceosome. Annu. Rev. Biochem. 89, 359–388 (2020).
Gooding, C. et al. A class of human exons with predicted distant branch points revealed by analysis of AG dinucleotide exclusion zones.Genome Biol. 7, R1 (2006).
Mercer, T. R. et al. Genome-wide discovery of human splicing branchpoints. Genome Res. 25, 290–303 (2015).
Taggart, A. J. et al. Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 27, 639–649 (2017).
Pineda, J. M. B. & Bradley, R. K. Most human introns are recognized via multiple and tissue-specific branchpoints. Genes Dev. 32, 577–591 (2018).
Gao, K. P., Masuda, A., Matsuura, T. & Ohno, K. Human branch point consensus sequence is yUnAy. Nucleic Acids Res. 36, 2257–2267 (2008).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535 (2019).
Lim, K. H., Ferraris, L., Filloux, M. E., Raphael, B. J. & Fairbrother, W. G. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl Acad. Sci. USA 108, 11093–11098 (2011).
da Costa, P. J., Menezes, J. & Romao, L. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int. J. Biochem. Cell Biol. 91, 168–175 (2017).
Group, P. T. C. et al. Genomic basis for RNA alterations in cancer. Nature 578, 129–136 (2020).
Gupta, A. K. et al. Degenerate minigene library analysis enables identification of altered branch point utilization by mutant splicing factor 3B1 (SF3B1). Nucleic Acids Res. 47, 970–980 (2019).
Cheung, R. et al. A multiplexed assay for exon recognition reveals that an unappreciated fraction of rare genetic variants cause large-effect splicing disruptions. Mol. Cell 73, 183 (2019).
**ong, H. Y. et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806 (2015).
Cheng, J. et al. MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 20, 48 (2019).
Pertea, M., Lin, X. & Salzberg, S. L. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 29, 1185–1190 (2001).
Rosenberg, A. B., Patwardhan, R. P., Shendure, J. & Seelig, G. Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 163, 698–711 (2015).
Jian, X., Boerwinkle, E. & Liu, X. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet. Med. 16, 497–503 (2014).
Riepe, T. V., Khan, M., Roosing, S., Cremers, F. P. M. & 't Hoen, P. A. C. Benchmarking deep learning splice prediction tools using functional splice assays. Hum. Mutat. 42, 799–810 (2021).
Soemedi, R. et al. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 49, 848–855 (2017).
Lin, H. et al. RegSNPs-intron: a computational framework for predicting pathogenic impact of intronic single nucleotide variants.Genome Biol. 20, 254 (2019).
Jagadeesh, K. A. et al. S-CAP extends pathogenicity prediction to genetic variants that affect RNA splicing. Nat. Genet. 51, 755 (2019).
Stenson, P. D. et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–581 (2003).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
Sherry, S. T., Ward, M. H. & Sirotkin, K. dbSNP – Database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9, 677–679 (1999).
Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017).
Adamson, S. I., Zhan, L. & Graveley, B. R. Vex-seq: high-throughput identification of the impact of genetic variation on pre-mRNA splicing efficiency. Genome Biol. 19, 71 (2018).
Amit, M. et al. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 1, 543–556 (2012).
Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Series B Stat. Methodol. 58, 267–288 (1996).
Leman, R. et al. Assessment of branch point prediction tools to predict physiological branch points and their alteration by variants. BMC Genomics 21, 86 (2020).
Lin, J. C., Fan, C. T., Liao, C. C. & Chen, Y. S. Taiwan Biobank: making cross-database convergence possible in the Big Data era. Gigascience 7, 1–4 (2018).
Song, K. et al. The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell 125, 453–466 (2006).
John, S. W. M. et al. Genetic decreases in atrial-natriuretic-peptide and salt-sensitive hypertension. Science 267, 679–681 (1995).
Chan, J. C. Y. et al. Hypertension in mice lacking the proatrial natriuretic peptide convertase corin. Proc. Natl Acad. Sci. USA 102, 785–790 (2005).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415 (2016).
Massaguer, A. et al. Characterization of platelet and soluble-porcine P-selectin (CD62P).Vet. Immunol. Immunopathol. 96, 169–181 (2003).
Baeza-Centurion, P., Minana, B., Valcarcel, J. & Lehner, B. Mutations primarily alter the inclusion of alternatively spliced exons.eLife 9, e59959 (2020).
Braun, S. et al. Decoding a cancer-relevant splicing decision in the RON proto-oncogene using high-throughput mutagenesis. Nat. Commun. 9, 3315 (2018).
Chiang, H. L., Wu, J. Y. & Chen, Y. T. Identification of functional single nucleotide polymorphisms in the branchpoint site. Hum. Genomics 11, 27 (2017).
Mikl, M., Hamburg, A., Pilpel, Y. & Segal, E. Dissecting splicing decisions and cell-to-cell variability with designed sequence libraries. Nat. Commun. 10, 4572 (2019).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Corvelo, A., Hallegger, M., Smith, C. W. J. & Eyras, E. Genome-wide association between branch point properties and alternative splicing.PLoS Comput. Biol. 6, e1001016 (2010).
Bonano, V. I., Oltean, S. & Garcia-Blanco, M. A. A protocol for imaging alternative splicing regulation in vivo using fluorescence reporters in transgenic mice. Nat. Protoc. 2, 2166–2181 (2007).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Cotto, K. C. et al. RegTools: Integrative analysis of genomic and transcriptomic data to identify splice altering mutations across 35 cancer types.Cancer Res. 80(16 Suppl), 2136 (2020).
Lorenz, R. et al. ViennaRNA Package 2.0.Algorithm Mol. Biol. 6, 26 (2011).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: A sequence logo generator. Genome Res 14, 1188–1190 (2004).
Ke, S. et al. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res. 21, 1360–1374 (2011).
Culler, S. J., Hoff, K. G., Voelker, R. B., Berglund, J. A. & Smolke, C. D. Functional selection and systematic analysis of intronic splicing elements identify active sequence motifs and associated splicing factors. Nucleic Acids Res. 38, 5152–5165 (2010).
Wang, Y., Ma, M., **ao, X. & Wang, Z. Intronic splicing enhancers, cognate splicing factors and context-dependent regulation rules. Nat. Struct. Mol. Biol. 19, 1044–1052 (2012).
Giudice, G., Sanchez-Cabo, F., Torroja, C. & Lara-Pezzi, E. ATtRACT – a database of RNA-binding proteins and associated motifs. Database (Oxford) 2016, baw035 (2016).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Tibshirani, R. et al. Strong rules for discarding predictors in lasso-type problems. J. R. Stat. Soc. Series B Stat. Methodol. 74, 245–266 (2012).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
Saito, T. & Rehmsmeier, M. Precrec: fast and accurate precision-recall and ROC curve calculations in R. Bioinformatics 33, 145–147 (2017).
Lin, J. C., Hsiao, W. W. W. & Fan, C. T. Transformation of the Taiwan Biobank 3.0: vertical and horizontal integration. J. Transl. Med 18, 304 (2020).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Shaun Purcell, C. C. PLINK. v.1.9 edn; www.cog-genomics.org/plink/1.9/ (2019).
Shaun Purcell, C. C. PLINK. v.2.0 edn; www.cog-genomics.org/plink/2.0/ (2019).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Ripley, B., Venables, W. & Ripley, M. B. Package ‘nnet’. R. package v.7, 3–12 (2016).
Acknowledgements
We thank M.-C. Tsai, Senior Scientific Editor at Cell, for constructive advice and editing the manuscript. We thank the Genomics Core of Institute of Molecular Biology (IMB), Academia Sinica, for performing the amplicon sequencing. We thank all members of IMB, particularly H.-J. Cheng, J.-Y. Leu, S.-C. Cheng and S.-H. Chen, for tremendous help and support. This work was supported by Career Development Award and Multidisciplinary Health Cloud Research Program of Academia Sinica (AS-CDA-108-M03 and AS-PH-109-01-3), Career Development Award of National Health Research Institute, Taiwan (NHRI-EX109-10908BC) and Excellent Young Scholar Research Grants and Ta-You Wu Memorial Award of Ministry of Science and Technology, Taiwan (MOST 109-2628-B-001-014-MY1 and 108-2118-M-001-013-MY5).
Author information
Authors and Affiliations
Contributions
H.-L.C., Y.-T.C., J.-Y.S., Y.-L.W. and C.-L.L. carried out experiments and analysis. H.-N.L., C.-H.A.Y., Y.-J.H., Y.-T.C. and C.-L.L. established the web server tool. Y.-T.H. supervised statistical analysis. H.-L.C., Y.-T.C., J.-Y.S. and C.-L.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Ana Fiszbein and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Sara Osman was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1
Analysis flow of the study.
Extended Data Fig. 2 Workflow of the library construction.
(a) Primers used in the overlap** PCR. (b) The procedure of overlap** PCR. In brief, oligo pools and the other parts of the splicing minigenes were amplified by 25 PCR cycles. The fragment containing the promoter and the first exon (PCR product 1) was stitched to the oligo pool (PCR product 2) by overlap** PCR using 20 amplification cycles. Then, the stitched product (PCR product 1+2) was further stitched with the fragment containing the 3rd exon and polyadenylation signal (PCR product 3) using 20 amplification cycles to obtain the final construct (PCR product 1+2+3).
Extended Data Fig. 3 Massively parallel splicing assay (MaPSy) showing high consistency between repeats.
(a) Spearman’s correlation between 4 RNA-seq total read counts after alignment. (b) Spearman’s correlation between 4 RNA-seq read count with spliced reads using GT-AG as splice sites. (c) Principle analysis for the spliced outcome of total RNA-seq read count. (d) Principle analysis for the spliced outcome of RNA-seq read count after GT-AG splice site filtration on spliced reads.
Extended Data Fig. 4 Individual WT/mt pair validation of the Massively parallel splicing assay (MaPSy).
Single minigene WT was transfected in HEK293T cells for splicing. The splicing outcome was examined by RT-PCR. Genomic coordinates, transcript ID, gene name and the corresponding introns were labeled accordingly. A representative experiment of three repeats is presented.
Extended Data Fig. 5 Characters of intronic 3’-end splicing variants.
(a) Significant splicing variants are significantly closer to the 3’ss. Identity of two distributions was examined by two-sided Kolmogorov-Smirnov test. (b) Mutation effect on splicing efficiency of variants of various pathogenic levels. n = 247 variants. (c) Genome-wide association between the number of intronic splicing regulatory elements near the 5’ss and the intron length, stratified by exon length, related to Fig. 2f. n = 704,953 introns. (d) Enrichment of add-AG variants in the significant splicing variants. (e) Differential exon-intron GC content for non-significant and significant add-AG variants, stratified by intron length, related to Fig. 3f. n = 281 add AG variants. **: P-value of two-sided Wilcoxon test between the non-significant and HC significant variants 9 × 10−4; * 0.01. The boxes in box plots represent medians (central line) and interquartile ranges (IQR; 25th to 75th percentile). The whiskers indicate ±1.5 × IQR from the box or the last data point within that and the dots show the outliers (b,c,e). (f) Preference of 3’ss with various 3’ss strengths was examined by 3’ss swap** assay with splicing minigenes. A representative experiment of three repeats is presented.
Extended Data Fig. 6 Sensitivity analysis of the generalized linear model that predicts splice-altering intronic mutations, related to Fig. 4.
(a) Segregation of intronic mutations into two models based on intronic location and AG addition, same as Fig. 4a. (b-d) The left-most presents the top 10 contributory factors predicting mutations in each category that affect splicing, same as Fig. 4b, d and f. Right next to the factors is the ‘variance inflation factor (VIF)’ that examines the collinearity of variables. VIF smaller than 5 is an indication of independence of variables without a collinear effect. Z-score of univariate GLM in the middle column shows the size of the marginal influence of each variable (without other variables in the model). The right-most figure shows the consistency of variable selection with 20 different random selections of training data.
Extended Data Fig. 7 Generalized linear model to synthesize predictors of splicing altering non-AG-creating intronic mutations and splicing efficiency without WT splicing efficiency.
(a) ROC curve of the non-AG model without WT splicing efficiency, similar to Fig. 4e. (b) Top 10 contributing factors to predict non-AG mutations affecting splicing without WT splicing efficiency, similar to Fig. 4d. (c) ROC curve of the 5’-end model without WT splicing efficiency, similar to Fig.4g. (d) Top 10 contributing factors to predict 5’-end mutations affecting splicing without WT splicing efficiency, similar to Fig. 4f. (e,g) Explanation power of each splicing efficiency model without WT splicing efficiency of (e) Novel AG mutations (g) non-AG mutations. The explanatory power of each model on the test dataset was estimated by Pearson’s correlation (two-tailed), R square, and RMSE (root-mean-square error). The gray area displays the 95% confidence interval for predictions from the linear model. (f,h) All contributing factors to predict (f) novel AG mutations and (h) non-AG mutations affecting splicing without WT splicing efficiency. An ‘alt’ factor refers to a canonical property in the context of sequence variation. A ‘△ (delta)’ factor refers to the difference of scores/motifs between the alt and WT sequence. A ‘novel’ factor refers to a new property associated with the novel 3’ss AG (f). More detailed descriptions of the factors can be found in Supplementary Table 2.
Extended Data Fig. 8 Intrinsic features of the BS and 3’ss regulate splice outcome.
(a) Genome-wide distribution of A-, C-, T- and G-branchpoints (bp) relative to the 3’ss. (b) Proportion of branchpoints supporting constitutive splicing, sorted according to relative distance to the 3’ss. (c) Minimum free energy of BS pairing with the U2 BS recognition region, represented by boxplots for each position. (d) PhyloP100 conservation level of bp relative to the 3’ss.
Supplementary information
Source data
Source Data Fig. 1
Unprocessed gels.
Source Data Fig. 7
Unprocessed gels.
Source Data Extended Data Fig. 4
Unprocessed gels.
Source Data Extended Data Fig. 5
Unprocessed gels.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chiang, HL., Chen, YT., Su, JY. et al. Mechanism and modeling of human disease-associated near-exon intronic variants that perturb RNA splicing. Nat Struct Mol Biol 29, 1043–1055 (2022). https://doi.org/10.1038/s41594-022-00844-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-022-00844-1
- Springer Nature America, Inc.