

Abstract
Highly multiplexed imaging holds enormous promise for understanding how spatial context shapes the activity of the genome and its products at multiple length scales. Here, we introduce a deep learning framework called CAMPA (Conditional Autoencoder for Multiplexed Pixel Analysis), which uses a conditional variational autoencoder to learn representations of molecular pixel profiles that are consistent across heterogeneous cell populations and experimental perturbations. Clustering these pixel-level representations identifies consistent subcellular landmarks, which can be quantitatively compared in terms of their size, shape, molecular composition and relative spatial organization. Using high-resolution multiplexed immunofluorescence, this reveals how subcellular organization changes upon perturbation of RNA synthesis, RNA processing or cell size, and uncovers links between the molecular composition of membraneless organelles and cell-to-cell variability in bulk RNA synthesis rates. By capturing interpretable cellular phenotypes, we anticipate that CAMPA will greatly accelerate the systematic map** of multiscale atlases of biological organization to identify the rules by which context shapes physiology and disease.
Similar content being viewed by others

Main
The wide availability of single-cell omics techniques has rapidly advanced our understanding of cell biology in health and disease1,2. Currently, there is a rapidly growing range of spatially resolved omics methods, which can quantify tens to hundreds of molecular species in single cells across large populations of cells or tissues, and at the same time show how these molecular species are spatially organized from the multicellular to the subcellular scale3,4,5. This combination of quantitative and spatial information across multiple scales holds enormous promise for understanding biological systems.
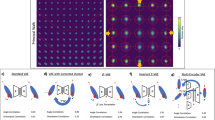
Cells in different states (for example, distinct cell cycle positions or disease states) or experimental conditions show changes in the relative abundance and subcellular localization of proteins and RNAs. From an analysis perspective, the challenge is to identify and quantify these changes directly from multiplexed image-based datasets in an unbiased manner, and thereby facilitate their biological interpretation. Previously, pixel clustering of multiplexed image data has been used to identify subcellular regions via similarity of their molecular profiles3,4. These approaches weigh all channels equally in clustering, therefore, when applied across cells from different experimental conditions they typically result in pixels from different conditions being identified as distinct4, even though they may represent the same subcellular region. As an extreme example, if an experimental treatment eliminates a single target protein (Fig. 1a), the reduction in intensity of the corresponding channel may be the largest difference between the high-dimensional pixel profiles of the two conditions. In this case, direct pixel clustering would identify independent sets of pixel clusters for each condition (Fig. 1b). Although this may be useful for qualitative identification of differences between conditions4, it does not enable quantification of changes in the internal organization of cells because it is difficult to compare the different sets of subcellular regions found in each condition (Supplementary Note 1).

a, Schematic showing perturbation-induced changes in channel intensity. b, Schematic of direct pixel clustering across experimental conditions leading to condition-dependent clusters. c, Schematic of CAMPA, showing how a cVAE conditioned on perturbation can learn a perturbation-independent latent space. Clustering this latent space identifies CSLs, enabling quantitative comparisons. d, Schematic of the 4i experiment and dataset dimensions. e, Fold-change in nuclear mean intensity in different perturbations compared with unperturbed cells, for all proteins with nuclear localization. P values show the significance of the perturbation effect on mean intensity, as determined using a mixed-effect model (Wald test, multiple testing correction using Benjamini–Yekutieli method). 5-EU represents 5-ethynyl uridine pulse labeling of nascent RNA (Methods). f, UMAP representation of pixels using either multiplexed pixel profiles (left) or cVAE latent space (right). Pixels from unperturbed cells, trichostatin A (TSA)-treated and triptolide-treated cells colored by perturbation. Data shown are the subset of pixel profiles used to derive the clustering (see Methods). g, Comparison of perturbation dependence of multiplexed pixel profiles, and VAE/cVAE latent space coordinates. Plots show balanced accuracy scores of binary logistic regression classifiers predicting perturbation from normalized multiplexed pixel profiles or latent representations. Accuracy of 0.5 indicates random chance (perturbation information absent from data). h, Example cells from each perturbation colored by clusters, along with a pie chart of relative abundance of clusters per perturbation. Left: Direct pixel intensity clustering (Leiden resolution, 1.2). Right: cVAE latent space clustering (CSLs) (Leiden resolution, 0.5). i, Comparison of perturbation dependence of direct clustering at different Leiden resolutions, and VAE and cVAE latent space clustering (CSLs). Plots show the coefficient of variation of the fraction of pixels assigned to each cluster in each perturbation. The boxplot summarizes results for all clusters with the number of clusters n shown above. Center line, median; box limits, upper and lower quartiles; whiskers, 1.5-fold the interquartile range; points, all data points.
Recently, deep learning-based segmentation models were used to segment cells and nuclei from multi-channel fluorescence microscopy images6,7. However, adapting these supervised methods to generate consistent segmentations of subcellular structures would require annotated training data from all conditions. Although self-supervised approaches alleviate the need for this time-consuming manual labeling8,9, they do not account for changing localizations of molecular species across perturbations nor do they enable quantification of these changes. To facilitate high-throughput quantitative analysis of subcellular organization, we therefore need approaches that can identify consistent subcellular landmarks despite condition-dependent, and possibly unanticipated, changes to abundance and/or relative localization of measured proteins and RNAs.
To achieve this, we have developed CAMPA (Conditional Autoencoder for Multiplexed Pixel Analysis), a deep learning framework based on conditional variational autoencoders (cVAEs)10. CAMPA uses a cVAE for unsupervised learning of condition-independent molecular profile representations to identify consistent subcellular landmarks (CSLs), that is, pixel clusters that are conserved across conditions. Using these landmarks to measure changes in molecular composition and spatial organization at the subcellular scale, CAMPA enables an interpretable comparison of conditions (Fig. 1c). CAMPA is an open-source python package with strong links to the single-cell transcriptomics analysis software, scanpy11, and its spatial extension, squidpy12. It enables high-throughput analysis of high-resolution multiplexed imaging datasets with GPU (graphics processing unit)-accelerated assignment of pixels to CSLs.
Here, we use CAMPA to derive a detailed map of subnuclear organization across different perturbations, directly from high-resolution iterative indirect immunofluorescence imaging (4i) (ref. 4) data. This shows how key proteins and protein states (for example, phosphoproteins and histone post-translational modifications) involved in transcription, chromatin, mRNA processing and nuclear export, as well as subnuclear organelles, change at the cellular and subcellular scale upon perturbation of various stages of messenger RNA metabolism. We find that the three aspects of cellular phenotypic information captured by CAMPA (cellular intensities, subcellular protein localizations and subcellular spatial organization) contribute unique information to characterize perturbations, indicating that CAMPA will be a powerful approach for cellular phenotypic screening. Finally, by capturing and quantifying interpretable cellular phenotypes at multiple scales, we demonstrate that the combination of 4i and CAMPA can uncover quantitative relationships across scales, from cell populations to subcellular organelles.
Results
CAMPA identifies consistent subcellular landmarks
In highly multiplexed image datasets, each pixel is represented as a multiplexed pixel profile: a one-dimensional vector containing the intensity of each marker at that spatial location. We developed CAMPA to identify consistent types of pixel profiles across different experimental conditions, even when some of the underlying channels change. CAMPA first learns a local, condition-independent representation of multiplexed pixel profiles and subsequently clusters the learned representations into CSLs (Fig. 1c). To learn a latent representation z, a cVAE is trained on an n × n neighborhood of the multiplexed pixel profiles x, together with a set of condition labels c for each pixel profile. Pixels are then grouped together by applying the Leiden algorithm13 on a k-nearest neighbor graph of the learned latent (pixel) representations. Because the cVAE model learns a conditional generative distribution \({p}_{\theta }({x|z},c)\) for the pixel profiles, the model is optimized to encode variation such as subcellular differences in intensity that occur across all conditions (and omit condition-specific information) in the latent representation z10,14, which results in less condition-dependent clustering of z (Fig. 1h,i). Within CAMPA, identified CSLs can be quantitatively compared in terms of their size, shape, molecular composition and relative spatial organization.
A key goal of perturbation experiments is to identify and quantify induced changes in cellular phenotypes. Here, we focus on how perturbation of various stages of RNA metabolism affects subcellular organization, by collecting a high-resolution (pixel size, 108 nm × 108 nm) 44-plex image dataset of 11,848 human epithelial cells (184A1) across six chemical perturbations, using 4i (ref. 4) (Fig. 1d). The perturbations target different pathways involved in RNA production and processing (histone deacetylation, trichostatin A (TSA); polymerase (Pol) I transcription, CX5461 (ref. 15); Pol II transcription initiation, triptolide16; Pol II transcription activation, AZD4573 (ref. 17); and mRNA splicing, meayamycin18). The proteins and post-translational modifications imaged (Supplementary Table 1) either play roles in RNA metabolism or are molecular markers of subcellular organelles (for example, nuclear speckles) or cellular states (for example, cell cycle stage, cell crowding). We observed changes in overall protein state abundances across all perturbations (Fig. 1e), confirming previous observations in other cell lines19. However, we also noticed perturbation-induced changes in the composition and relative spatial organization of membraneless nuclear organelles involved in RNA metabolism, such as nuclear speckles, promyelocytic leukemia (PML) bodies and the nucleolus. This dataset therefore provides an ideal use-case for the CAMPA framework to generate novel insights into relationships between RNA metabolism and subcellular organization.
To quantify these changes, we initially focused on analyzing the approximately 100 million nuclear pixels for the 34 markers that localized to the nucleus (Extended Data Fig. 1 and Supplementary Tables 2, 3). We applied CAMPA cVAE training and clustering to these data using cell cycle stage (labeled independently of CAMPA19,20) and perturbation condition as categorical condition labels. As expected, we found that multiplexed pixel profiles were highly perturbation dependent when plotted using UMAP (uniform manifold approximation and projection) embedding21, while the cVAE latent representations appeared to have overlap** distributions (Fig. 1f). To verify the condition independence of the latent representation, we used binary linear classifiers trained to distinguish pixels from perturbed and unperturbed cells based on their latent representations. These classifiers were often not better than random chance (median accuracy, 0.53; minimum, 0.50; maximum, 0.60). In contrast, classifiers based on multiplexed pixel profiles reached a median accuracy of 0.87 (minimum, 0.52; maximum, 0.98). The VAE model without conditioning was not able to generate condition-independent latent spaces (median accuracy, 0.72; minimum, 0.51; maximum, 0.97), indicating that explicit use of conditioning is necessary in CAMPA (Fig. 1g and Extended Data Fig. 2a). To investigate the importance that the cVAE places on the condition, we used integrated gradients22, which showed that for channels with perturbation-specific intensity changes, the cVAE places increased importance on the condition input (as opposed to the latent representation) for modeling these channels (Extended Data Fig. 4d,e). We also optimized the input neighborhood size to improve cVAE latent space robustness to single-pixel noise, which often occurs in microscopy imaging. For our data, a 3 × 3 neighborhood was optimal (Supplementary Fig. 1d).
To accelerate latent space clustering and to enable interactive clustering on a standard workstation, we clustered a subsample of pixels (150,000 pixels) and then projected resulting clusters to all pixels using the 15 nearest neighbors. This resulted in 10 clusters. Cluster stability was not significantly influenced by a different random subsample nor by increasing or decreasing the number of samples used for the clustering by a factor of two (Supplementary Fig. 1a,b). Because all conditions are considered together, any cluster instability does not affect the ability to quantitatively compare cells across conditions. For comparison with previous approaches, we also directly clustered pixels using their multiplexed pixel profiles4. Whereas intensity space clusters were enriched in different perturbations (Fig. 1h and Extended Data Fig. 2b), latent space clusters were evenly distributed across perturbations (Fig. 1h and Extended Data Fig. 2c). To quantify the perturbation specificity of clusters, we computed the median coefficient of variation of the fraction of pixels assigned to each cluster across perturbations. The median coefficient of variation of the latent space clustering is 0.24 (minimum, 0.08; maximum, 0.61), indicating that clusters have a similar relative abundance in different perturbations, whereas direct pixel clustering at similar resolution results in a median coefficient of variation of 0.57 (minimum, 0.09; maximum, 2.62) (Fig. 1i). In addition, despite differences in intensities of some 4i markers across different cell cycle phases (for example, PCNA (proliferating cell nuclear antigen), pRB1), the inclusion of cell cycle as a condition in CAMPA reduced the cell cycle dependence of the latent representations (median accuracy of pairwise binary classifiers of latent space/pixel profiles, 0.58/0.67), which resulted in latent space clusters being assigned consistently across cell cycle stages (median coefficient of variation across cell cycle stages of latent space clustering/direct pixel clustering, 0.11/0.21) (Extended Data Fig. 3). We therefore name these cVAE latent space clusters ‘consistent subcellular landmarks’ (CSLs) and use them in the following to analyze the impact of perturbations on subcellular organization.
To enable biological interpretability of quantitative comparisons between cells, we annotated CSLs with the names of known subcellular structures (see Methods) (Fig. 2a). To facilitate this optional step in the CAMPA workflow and to avoid mis-annotations, automated annotation proposals can be obtained by querying the Human Protein Atlas (https://www.proteinatlas.org/)23 database. The annotation resulted in assignment of the 10 original CSLs to seven annotated CSLs (Nucleolus, Nuclear speckles, PML bodies, Cajal bodies, Nucleoplasm, Nuclear periphery and Extra-nuclear (outside the nucleus)) (Fig. 2d–i), by merging four original CSLs into the Nucleoplasm CSL (Extended Data Fig. 4a). These annotations are consistent with automatic annotations proposed by the Human Protein Atlas database (Extended Data Fig. 4b). In the following we refer to these annotated CSLs simply as CSLs. To quantitatively validate CSL annotations, we performed two manual segmentations of nuclear speckles and two manual segmentations of PML bodies using state-of-the-art pixel classifiers24 (Extended Data Fig. 5). These were based only on single-channel intensities of canonical markers for these membraneless organelles (SON and SRRM2 for nuclear speckles and SP100 and PML for PML bodies). We quantitatively compared these manual segmentations with their respective CSLs using the F1-score (a measure of similarity) and found that CSL-derived nuclear speckles were as similar to the manual segmentations (\({F}_{1({CSL|SON})}=0.963\pm 0.006\), \({F}_{1({CSL|SRRM}2)}=0.967\pm 0.006\), mean ± s.d. between conditions) as the different manual segmentations are to one another (\({F}_{1({SRRM}2{|SON})}=0.964\pm 0.007\)) (Extended Data Fig. 5). F1-scores were similarly high for PML bodies.

a, UMAP representation of pixels using their cVAE latent representations generated in CAMPA, colored by CSL. b, Example nucleus showing the spatial distribution of CSLs. c, Relative mean intensity of each channel in each annotated CSL (see Extended Data Fig. 4a for all 10 Leiden clusters). Heatmap z-scored by column to show the relative localization of each channel across CSLs. The black-outlined boxes are highlighted in d–i. d–i, Example 4i channels that are enriched or depleted in the identified CSLs, shown together with CSLs. See c for the distribution of channels across the CSLs. Scale bar, 5 µm.
We therefore conclude that CAMPA enables consistent identification and annotation of subcellular landmarks across perturbations and cell cycle stages. This contrasts with previous direct pixel clustering approaches, which often identify different clusters for the same subcellular organelle in different conditions or cell cycle stages. Unlike for manual segmentation of subcellular structures, when using CAMPA to identify CSLs there is no need to pre-define markers of certain landmarks in advance, because the cVAE uses all channels that are consistent across perturbations to define the latent space. This may ultimately enable identification of novel landmarks defined by higher-dimensional combinations of different channels. Importantly, the cVAE learns to remove condition-specific information from channels that show characteristic changes in intensity between conditions when generating the latent space and the CSLs. Naturally, as shown in the following, these channels can then be used to compare the effects of, and differences between, perturbations when aggregated on the CSLs.
Uncovering perturbation-induced subcellular landmark changes
To quantify subcellular changes in abundance of markers across the six perturbations, we calculated the mean intensity of each marker in each CSL per cell. We then computed the fold-change for a particular condition compared with unperturbed cells, across all CSL–channel combinations, as well as the fold-changes in the size (number of pixels) of each CSL (Supplementary Fig. 2a,b). Unlike direct pixel clustering approaches3,4, in which conditions are compared by identifying pixel classes that change abundance between conditions (Extended Data Fig. 6 and Supplementary Note 1), CAMPA compares molecular abundances across landmarks that are consistently found in both conditions (CSLs). This naturally extends traditional quantification of overall cellular abundance changes (Fig. 1d) to the subcellular scale. Focusing on meayamycin, which perturbs mRNA splicing18, CAMPA identified a set of markers that were uniformly depleted across the nucleus, and an overall increase in the size of nuclear speckles (Supplementary Fig. 2b). To investigate relocalization of proteins (rather than overall changes in abundance), we normalized intensity fold-changes in each CSL by their corresponding whole-nucleus fold-changes (Fig. 3a and Supplementary Fig. 2c). This showed that the relative size of nuclear speckles increases upon meayamycin treatment, and that their molecular composition changes: they become significantly enriched in cytoplasmic poly(A) binding protein 1 (PABPC1) (Fig. 3d) and depleted in POLR2A-S2P (a marker of actively transcribing RNA polymerase II) (Fig. 3e). PABPC1 relocalization to nuclear speckles was observed previously25. POLR2A-S2P is typically distributed throughout the nucleoplasm with slight enrichment in nuclear speckles (Fig. 2c)Methods). Scale bars: a, 20 µm; e–g, 20 μm.
This analysis shows that CAMPA generalizes to a higher level of multiplexing and can identify CSLs not only across conditions with different molecular profiles but also across different CSL sizes. Morphological properties of CSLs on a per-cell basis such as count and area can be used to compare and interpret changes in scaling behavior between conditions.
Linking cellular heterogeneity to subcellular reorganization
Finally, we use CAMPA to study how subcellular properties vary within cell populations, to examine its potential in uncovering links between subcellular properties and cellular states. Rates of RNA production are heterogeneous in cell populations19,38 and can be measured by RNA metabolic labeling with 5-ethynyl uridine39. Nuclear 5-ethynyl uridine intensity quantifies the amount of nascent RNA synthesized during a 5-ethynyl uridine pulse at the single-cell level (Fig. 6a). To examine how differences in bulk RNA production are related to subcellular changes, we considered control cells (scrambled siRNA) from the CAMPA model trained on entire HeLa cells (Fig. 5) and binned these into either ‘low’ (lower quartile) or ‘high’ (upper quartile) RNA synthesis, using mean nuclear 5-ethynyl uridine intensity (Extended Data Fig. 9a). Examination of intensity fold-changes for each channel–CSL combination between these groups revealed changes in overall nuclear concentration of POLR2A and other proteins and protein states related to RNA synthesis (Extended Data Fig. 9b), as previously observed19. Focusing on the subcellular level, we observed that PML bodies showed a change in the relative molecular composition of PML and SP100, the two markers of PML bodies used in this experiment. In cells with low RNA synthesis, PML bodies were enriched in PML, while in cells with high RNA synthesis, PML bodies were enriched for SP100. These changes are difficult to observe in overall (all) or whole-nucleus (Nucleus (combined)) CSLs, demonstrating the importance of quantifying this at the subcellular scale. These trends were recapitulated across the full range of 5-ethynyl uridine intensities (Fig. 6b), and were observed in the G1, S and G2 phases of the cell cycle (Extended Data Fig. 9c). PML bodies have previously been implicated in transcriptional regulation40, however, their molecular composition has not been linked to global changes in transcriptional output of single cells. Examining images directly revealed heterogeneity in PML body composition, both between and in cells (Fig. 6c). Specifically, cells with low RNA synthesis had PML bodies lacking SP100, while high RNA synthesis cells had PML bodies lacking PML. Classically, these bodies are defined as having both SP100 and PML41. Detection of these nuclear bodies based only on PML or on SP100 (univariate) would have not assigned all these pixels as PML bodies, highlighting a key difference between CAMPA and univariate approaches. It is important to note, however, that, given that we did not use 5-ethynyl uridine intensity as a condition in the cVAE training, we would expect to see these unique pixel combinations annotated as different CSLs at higher clustering resolution (Extended Data Fig. 9d). These results demonstrate that CAMPA can be used not only to reveal changes between perturbations but also to uncover links between global properties of cells and their subcellular organization.

a, Schematic of RNA metabolic pulse labeling with 5-EU39. b, Mean intensity of PML and SP100 in PML bodies as a function of mean nuclear 5-EU intensity (RNA synthesis rate) relative to the mean across all cells. All cells binned by 5-EU. Bins with less than 10 cells were omitted. Error bars show 95% confidence intervals for mean (obtained using bootstrap**; n = 500). Fit lines show LOESS regression of binned data with the shaded region representing the 95% confidence interval. The upper panel shows the number of cells in each bin (total n = 2,301 (scrambled siRNA, see Supplementary Table 5)). c, Example images comparing two S-phase cells in states of high and low RNA synthesis. Arrows highlight a PML-only PML body (low RNA synthesis, left) and an SP100-only PML body (high RNA synthesis, right). The outlines of PML bodies derived from PML body CSL were dilated by 9 pixels for visualization purposes. Scale bar, 10 µm.
