

Abstract
Invasive Staphylococcus aureus infections are common, causing high mortality, compounded by the propensity of the bacterium to develop drug resistance. S. aureus is an excellent case study of the potential for a bacterium to be commensal, colonizing, latent or disease-causing; these states defined by the interplay between S. aureus and host. This interplay is multidimensional and evolving, exemplified by the spread of S. aureus between humans and other animal reservoirs and the lack of success in vaccine development. In this Review, we examine recent advances in understanding the S. aureus–host interactions that lead to infections. We revisit the primary role of neutrophils in controlling infection, summarizing the discovery of new immune evasion molecules and the discovery of new functions ascribed to well-known virulence factors. We explore the intriguing intersection of bacterial and host metabolism, where crosstalk in both directions can influence immune responses and infection outcomes. This Review also assesses the surprising genomic plasticity of S. aureus, its dualism as a multi-mammalian species commensal and opportunistic pathogen and our develo** understanding of the roles of other bacteria in sha** S. aureus colonization.
Similar content being viewed by others


Introduction
Staphylococcus aureus is a frequent colonizer of the human population and one of the foremost opportunistic bacterial pathogens of humans, causing major morbidity and mortality globally1. Any clinician, especially those working in infectious diseases, will attest to the frequency, complexity and the potential catastrophic nature of invasive infections caused by this pathogen. Although on a global scale human colonization and infection with S. aureus are forefront of mind, the organism has a long evolutionary history as a multihost opportunistic pathogen. S. aureus colonizes approximately 20–30% of humans persistently in the nose2 and frequently in other sites such as the skin, throat, axillae, groin and intestine3. The interplay between the potential pathogen and the host microbiota is being explored with increasing depth and complexity, highlighting roles for other commensals in sha** S. aureus colonization4. Colonization is harmless but it is a risk factor for develo** subsequent infections (often caused by the colonizing strain5), which can range from mild skin and soft tissue infections to serious invasive infections, including osteomyelitis and septic arthritis, bacteraemia or septicaemia, pneumonia and endocarditis1. S. aureus infections can be acute, recurrent or chronic and persistent. To be best placed to develop effective therapies and clinical responses to these infections, we need to understand the complex pathophysiological relationships between S. aureus and its hosts6.
The breadth of immune evasion mechanisms in S. aureus reflects the longstanding nature of the interaction of bacterium with humans, characterized by persistent colonization and intermittent invasive infections. S. aureus produces a large array of virulence and immune evasion factors that hinder the human immune response7,8. Neutrophils represent the major defence against staphylococcal infections. Staphylococcal factors that perturb adaptive B cell and T cell responses and diminish protective immunity are well described8. Factors subverting innate immune response also dominate, such as inhibition of neutrophil chemotaxis and killing, inhibition of complement activation and phagocytosis, killing of host cells and staphylococcal agglutination6,8. Reflecting the presence of factors inhibiting adaptive immune responses, infection with S. aureus does not elicit a protective immune response, meaning that recurrent infections are common throughout life9,10. Decades of vaccine studies with promising preclinical results have failed to translate into effective vaccines for use in human, including the failure of two major phase III clinical trials to demonstrate a benefit11. The adaptation of some S. aureus clones to the human host means that preclinical animal models are limited in utility11.
In the colonization state, S. aureus is part of a polymicrobial community that primarily includes other staphylococci, corynebacteria and propionibacteria12,13. The roles of non-pathogenic commensals and interspecies interactions that influence S. aureus colonization and disease are largely unexplored14. However, advances in genomics-enabled microbial community analyses and metabolomics provide an opportunity to delve into these interactions (Supplementary Box 1 and Supplementary Fig. 1). In vivo S. aureus biofilms are more typically a monomicrobial community, in which surface-exposed proteins, referred to as microbial surface components recognizing adhesive matrix molecules (MSCRAMMs)15, initiate the attachment to surfaces, further consolidated by the subsequent release of polysaccharides, proteins and extracellular DNA, forming an extracellular matrix encasing the bacteria. Biofilm-associated infections, which have a propensity to chronicity through increased antimicrobial resistance and immune evasion, are frequently linked to implanted devices and contribute to the pathogenesis of S. aureus infections in native tissues15,16. The mechanisms of immune evasion afforded by S. aureus biofilms have not been clearly elucidated, but are thought to be in part caused by the masking of pathogen molecular signatures17.
Antimicrobial resistance acquired through acquisition of mobile genetic elements or chromosomal mutations has further complicated staphylococcal infections and therapeutics18. Although penicillin initially revolutionized the treatment of serious S. aureus infections, resistance through the acquisition of the β-lactamase encoding gene blaZ was soon widespread19. The acquisition of resistance to anti-staphylococcal penicillins through the mecA gene has resulted in the global dissemination of diverse lineages of methicillin-resistant S. aureus (MRSA), now considered a global public health threat19. Glycopeptides became the mainstay of therapy for invasive MRSA infections, but reduced susceptibility through adaptive mutations further hampered therapeutic efforts20, which results in the move to using ‘last-line’ antimicrobials such as daptomycin, and combination therapies19,20. Pathogen evolution in cases of antimicrobial resistance and persistent human infections occurs in the milieu of the host environment, which imparts an additional selective pressure potentially selecting for not just drug resistance but also immune escape21 (Supplementary Box 2 and Supplementary Fig. 2). The advent of accessible, high-throughput pathogen genomics technologies has revolutionized the study of staphylococcal resistance and adaptation, and the investigation of complex phenotypes linked to antimicrobial resistance, such as small colony variant (SCV) S. aureus22. The genomes of tens of thousands S. aureus isolates have been sequenced and are publicly available, and we are at an exciting stage in the utilization of this incredible resource for further understanding the pathogen23. Early use of the technology enabled the tracking of mutations that evolve during persistent clinical infections, which revealed an unexpected capacity for the pathogen to adapt to complex antimicrobial exposures and the host environment24,25, and uncovered S. aureus genetic heterogeneity that can exist within host26 (Box 1).
In addition to the growing list of newly discovered immune-modulating molecules, recent advances in our understanding of S. aureus pathogenesis and disease are highlighting the roles of biofilm formation, immunometabolic host interplay and immune evasion phenotypes (such as the SCV) to avoid host clearance. Added to this, S. aureus genomic studies are revealing a surprisingly high mutagenic potential that also materially contributes to immune evasion phenotypes. Finally, assessing bacterial adaptation processes occurring during switching between different vertebrate hosts is providing insights into host-specific disease mechanisms. In this Review, we examine and integrate recent key advances in understanding the mechanisms that S. aureus uses to cause infections. We explore our understanding of S. aureus immune evasion molecules and the role of biofilms, immunometabolism and S. aureus avoidance mechanisms of trained innate immunity. We also investigate S. aureus host specificity and adaptation and summarize current knowledge of the interplay between colonizing S. aureus and other bacteria with a summary of our emerging knowledge of S. aureus genomic plasticity, drug resistance and persistence. A detailed dissection of host responses, vaccines and other therapeutics is not covered in this Review.
Role of biofilms and immunometabolism
In addition to the host evasion factors described in the previous section, biofilm formation is another mechanism that contributes to the pathogenesis of S. aureus76. Biofilms on biomedical devices, such as catheters and medical implants, or on host tissues such as wounds or heart valves cause notoriously difficult-to-treat infections77. By embedding bacteria in an extracellular matrix consisting of proteins, polysaccharides and extracellular DNA, biofilms confer protection against antimicrobial therapeutics and host immune clearance77. Nonetheless, secretion of factors including haemolysins, nucleases and PSMs are also important mediators of host evasion during the biofilm lifestyle and could shift immune responses towards an anti-inflammatory state78,79.
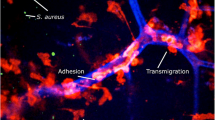
The contribution of the leukocidins Panton–Valentine leukocidin (PVL) and S. aureus γ-haemolysin (HlgAB) to bacterial survival in biofilms was demonstrated using a porcine wound model17. High-level secretion of PVL and HlgAB in biofilms elicited the formation of NETs by neutrophils (Fig. 1). Although NETs trapped and killed planktonic bacteria, they had no activity against S. aureus in biofilm17. S. aureus nuclease (Nuc) degraded the DNA content of NETs, which is likely to have promoted the dispersal of staphylococcal cells from the biofilm, increasing the risk of metastatic infections76. Additionally, LukAB has been shown to contribute to evasion of phagocyte-mediated killing of S. aureus in biofilm76.
S. aureus biofilms skew the host immune response towards an anti-inflammatory state, thereby promoting bacterial persistence instead of clearance (Fig. 3). This is evidenced by the recruitment of myeloid-derived suppressor cells (MDSCs)80,81,82,83 and the polarization of anti-inflammatory M2-like macrophages84. MDSCs are typically prominent in tumour microenvironments and notorious for their immunosuppressive effect, inhibiting T cell activation and promoting tumour progression (reviewed elsewhere85). Analysis of purified MDSCs recovered from S. aureus biofilm-associated orthopaedic infection revealed increased expression of anti-inflammatory mediators such as arginase 1 and IL-10 (ref. 82). Furthermore, depletion of MDSCs promoted bacterial clearance82.

The left panel shows metabolic reprogramming, characterized by increased glycolysis, in macrophages infected with live planktonic bacteria or stimulated with pathogen-associated molecular patterns such as lipopolysaccharide (LPS) recognized by pathogen recognition receptor (PRR). This skews the macrophage towards a pro-inflammatory phenotype (that is, the production of the pro-inflammatory cytokine IL-1β), which promotes bacterial clearance. The right panel shows two different strategies through which staphylococcal biofilms evade innate immunity. These include either rewiring host metabolism or epigenetic changes. S. aureus biofilm, which is induced by host-derived itaconate, promotes mitochondrial oxidative phosphorylation (OXPHOS) in macrophages and an anti-inflammatory phenotype. The mechanism remains unknown (indicated by the question mark). Lactate produced by S. aureus biofilms is transported into macrophages and myeloid-derived suppressor cells (MDSCs) through the monocarboxylate transporter 1 (MCT1), inhibits histone deacetylase 11 (HDAC11) and promotes IL-10 production. Ac, acetylation; CoA, coenzyme A; TCA, tricarboxylic acid.
Typically, macrophages become activated on recognition of bacteria, with the ensuing inflammatory response driven by major cellular metabolic changes, a process termed immunometabolism86. These changes include increased host cell glycolysis, impaired tricarboxylic acid (TCA) cycle and mitochondrial oxidative phosphorylation (OXPHOS), all resulting in the cellular accumulation of succinate and itaconate, which are two TCA metabolites87,88. Succinate is a pro-inflammatory metabolite that stabilizes the transcription factor, hypoxia-inducible factor 1𝛼, which enhances the production of the pro-inflammatory cytokine IL-1β; whereas itaconate is an anti-inflammatory metabolite that restores homeostasis following succinate-mediated inflammation87,88. Itaconate exerts its immunomodulatory role by alkylating cysteine residues and inhibiting several targets including the NLRP3 inflammasome, glycolytic enzymes and succinate dehydrogenase (SDH) mitochondrial complex II88,89,90. Interestingly, rather than stimulating inflammatory macrophages, S. aureus biofilms induce the polarization of macrophages into an anti-inflammatory state, with a metabolic bias towards increased OXPHOS and decreased aerobic glycolysis84 (Fig. 3). Importantly, the delivery of nanoparticles containing oligomycin, which is an OXPHOS inhibitor, in a mouse model of prosthetic joint infection resulted in the polarization of inflammatory monocytes and enhanced bacterial clearance. This demonstrates the potential of immunometabolic therapy in treating persistent staphylococcal infections (Box 3).
The increased expression of anti-inflammatory IL-10 by monocytes and MDSCs recruited to biofilms has been attributed to staphylococcal lactate, a metabolite that inhibits histone deacetylase 11 (HDAC11), thus enhancing IL-10 gene transcription91. This example highlights the crucial contribution of active bacterial metabolism and its crosstalk with host metabolic activities in determining the outcome of infection. Although mechanisms promoting immune evasion in biofilm-associated infections are being revealed92, the factors driving S. aureus biofilm formation in vivo are poorly defined. A recent study exploring the role of host immunometabolism during S. aureus pulmonary infection identified the inhibitory effect of the host metabolite itaconate on bacterial glycolysis as a biofilm-promoting cue93.
Itaconate is produced by the enzyme ACOD1 (also known as IRG1) in myeloid cells, particularly in response to lipopolysaccharide stimulation94. Of note, despite lacking lipopolysaccharide, S. aureus induces the production of host itaconate via the stimulation of mitochondrial oxidant stress93. Itaconate in turn inhibits the staphylococcal glycolytic enzyme aldolase, mirroring one of its interactions in mammalian cells90. Given the preferential reliance of S. aureus on glucose consumption and glycolysis for growth from carbon catabolite repression, the inhibition of glycolysis by itaconate rewires S. aureus metabolism, directing carbon flux towards biofilm formation and a sessile lifestyle. In an infection context, the consequence of metabolic adaptation for biofilm formation was revealed in a longitudinal study of S. aureus isolates colonizing the airways of a patient with cystic fibrosis over 15 years. These isolates displayed increasing production of biofilms in the presence of itaconate, consistent with the role of this metabolite in driving bacterial adaptation via biofilm formation93. Although the mechanism through which itaconate promotes persistent staphylococcal pulmonary infection and its clinical relevance requires further exploration, this study provides an intriguing insight into the immunometabolic pressures that support pathogen immune evasion adaptive mechanisms, akin to antibiotic selective pressure.
S. aureus small colony variants
The subversion of host immunometabolism by pathogens profoundly influences the infection outcome, as highlighted in the previous section. The compounding effect of accumulated metabolites on the epigenetic landscape of immune cells adds a further level of complexity to host–pathogen interactions. This is best exemplified by trained immunity or ‘innate immune memory’, which refers to increased innate immune protection against a secondary challenge following primary infection. Trained macrophages undergo substantial metabolic changes including the hallmark increase in glycolysis, and they accumulate metabolites that induce epigenetic rewiring of cellular function95. Accumulated fumarate inhibits KDM5 histone demethylases, which promotes trimethylation at the fourth lysine residue of the histone protein H3 (H3K4me3) at the promoters of pro-inflammatory cytokine genes such as those encoding tumour necrosis factor and IL-6, thus inducing their rapid production on restimulation and secondary infection.
In a mouse skin infection model, priming by previous infection with wild type S. aureus USA300 isolate LAC reduced skin lesion severity and bacterial burden on secondary infection. This protection was highly localized and mediated by macrophages96,97. Importantly, adoptive transfer of wild type S. aureus-primed macrophages into naive mouse skin afforded protection in vivo. However, priming by an S. aureus SCV prototype, ∆hemB, or a host-adapted isolate from a patient with atopic dermatitis did not confer protection from secondary infection98,99. SCVs, which are often associated with chronic infections, were first characterized in 1995 as phenotypically distinct from normal S. aureus colonies100. SCVs are slow-growing, have pinpoint colony size and may carry mutations in genes associated with electron transport chain components such as menadione and haeme22. As a result of these mutations, SCVs often have altered bacterial metabolism with decreased TCA cycle or OXPHOS activity and increased glycolysis to meet their energy requirements101. The ability of SCVs to abrogate trained immunity and cause recurrent infection was mediated by increased expression of fumC, which encodes the enzyme fumarate hydratase to degrade fumarate99. Increased fumC expression was also observed in S. aureus isolates from patients with atopic dermatitis and cystic fibrosis98,102. The blunting of immunity by host-adapted S. aureus isolates remains to be explored in the persistently colonized organs, such as the lungs of patients with cystic fibrosis. Genomics-enabled studies are providing further insights into SCV genetic mechanisms such as chromosomal rearrangements103. Wider uptake of such approaches is likely to accelerate the discovery of other bacterial genomic modifications occurring in SCVs and arising in response to immune selective pressures and promoting persistent infections (Supplementary Box 2 and Supplementary Fig. 2).
S. aureus host specificity and adaptation
Understanding host adaptation is providing insights into host-specific immune evasion mechanisms and by extension enhances our understanding of disease at the species level. It is well recognized that S. aureus is a multispecies pathogen, able to cause disease in humans and livestock. The diseases in livestock include mastitis in cows, goats, sheep and rabbits, skin infections in pigs and rabbits and invasive infections in chickens104. Advances in agricultural practices are increasing transmission risks between humans and livestock. For instance, the threat to human health from livestock-associated S. aureus has been well described for the pig-associated clones, especially CC398 (ref. 105) as well as ST9, ST5 (ref. 106) and ST93 (ref. 107); however, key immune evasion factors (scn, chp, sak, sea, which are encoded by the immune evasion complex) are observed in isolates from humans but not from pigs. Other clones from livestock can also spread in humans108 and exemplified by the recent description of the emergence of MRSA in non-human mammals, in this case European hedgehogs, which subsequently spread to livestock and humans, before the use of methicillin as an antibiotic109.
Host adaptation can be shaped by genetic drift (a process of neutral diversification) or adaptive evolution, whereby beneficial mutations are selected or deleterious mutations removed in the new host110. Genetic drift is typically associated with reduction of the effective population size as it occurs in ‘bottlenecks’111. For example, in the transition from colonizing to invasive pathogen, only a few cells of a more diverse S. aureus colonizing population survive within a macrophage or other phagocytic cell (bottlenecks) and then subsequently expand to cause invasive disease112. By contrast, adaptive evolution results in an increase in frequency of the beneficial variant. It is likely that both have a role during infection and adaptation113. Combining comparative population genomics including both human and animal isolates with functional genomic studies will illuminate the staphylococcal drivers of host adaptation and determine the host specific factors that support bacterial survival114. Such information will guide the design of strategies that prevent the emergence of new pandemic clones and cross-species transmission.
Over the past decade, genomics studies have provided a comprehensive map** of S. aureus accessory genome elements that are specific to certain host species and enriched the understanding of S. aureus host adaptation115, revealing that immune evasion factors targeted to certain hosts were specifically produced by host-adapted S. aureus lineages. Larger scale analyses of S. aureus population genomics across multiple host species and humans demonstrated that the latter were the major donors for S. aureus host-switching events, and that cows represent a potential reservoir of new epidemic clones in humans104. Understanding the gene flow in different ecological niches highlights host-specific gene repertoires that are linked to host adaptation, with S. aureus accessory genome elements from isolates from the same host species highly correlated even across diverse S. aureus clonal complexes104. By contrast, core genome evolution can be traced through the assessment of adaptive mutations that result from diversifying selection, with some biological pathways found to be under positive selection in humans (Fig. 4 and Supplementary Fig. 3). Antimicrobial resistance elements are another component of the accessory genome which can reveal unique host species patterns. Unsurprisingly, human isolates are enriched for elements that provide resistance to antimicrobials used in humans such as β-lactams, whereas tetracycline and antiseptic resistance are significantly associated with pig isolates104.

a, Host-switching modelling of Staphylococcus aureus between species identified humans as a major transmission hub104. Overlayed are reported host-specific mobile genetic elements (MGEs) in pigs118,119, cows119,120, small ruminants121,122, horses123, birds119 and hedgehogs109; and their major associated clonal complexes (CCs). MGEs belonging to the non-dominant S. aureus lineages in pigs and cows have been reported, these CCs and sequence types (STs) are denoted in superscript. S. aureus infecting rabbits has no unique MGEs. A single nucleotide polymorphism (C338A) in chromosomal gene, dtlB, causes a non-synonymous mutation required for infection of rabbits by CC121 (see Supplementary Fig. 3c for all CC121 rabbit adaptations). No unique S. aureus MGEs have been described in rodents or carnivora. Globally disseminated, multihost lineage CC398 has been reported in rodents and carnivora, but no CCs are recognized as dominant in these species (see Supplementary Fig. 3d for the mechanism of broad host distribution for CC398). Human S. aureus strains possess the ϕSa3 prophage (β-haemolysin-converting phage) encoding a human-specific evasion cluster that integrates into hlb, causing loss of β-toxin production. Many animal-adapted S. aureus strains have lost ϕSa3, resulting in restored β-toxin production and increased fitness. Linewidth represents frequency of host jumps. b, Function of S. aureus genes carried on animal-adapted MGEs. S. aureus superantigens are virulence factors that cause nonspecific T cell activation and massive cytokine release. This excessive activation of the host immune system paradoxically promotes infection; antibiotic resistance is conferred by multiple resistance determinants; species-adapted leukodicins, encoded by luk, cause host neutrophil lysis. von Willebrand-binding protein (vWbp) causes host-specific plasma coagulation; adhesion protein (Bap) promotes S. aureus adhesion to bovine mammary mucosa and biofilm production resulting in colonization, contributing to the pathogenesis of bovine mastitis. Putative adhesin, LPXTG-surface protein promotes colonization (see Supplementary Fig. 3a for bovine-specific S. aureus adaptions); staphylococcal complement inhibitor (SCIN) encoded by scn blocks the activation of the host complement system; Avian-specific scpA encodes the thiol protease staphopain A, which is associated with enhanced pathogenicity through the inhibition of host neutrophil activation and chemotaxis (see Supplementary Fig. 3b for S. aureus CC5 adaptations in chickens). #Putative adhesin. ϕSabov-vSa, bacteriophage ϕ-S. aureus bovine in genomic island v-S. aureus; LPXTG, conserved peptide motif in surface linked proteins cleaved between the threonine and glycine residues and covalently attached to cell wall peptidoglycan; SCCmecC, staphylococcal cassette chromosome methicillin C resistance gene; TSST-1, toxic shock syndrome toxin-1; vSaα, genomic island v-S. aureus-α.
The exploration of host-specific adaptation in experimental models could provide robust validation of genetic signatures that have been identified by population genomics analyses. An exemplar of a targeted approach is the study of ST121 S. aureus, which causes serious infections in both human and rabbit hosts. Bayesian modelling of genomic data suggests a human-to-rabbit host jump about 50 years ago, with genome diversification between host species since then116. Although no specific mobile elements were identified in the genome of rabbit-associated S. aureus ST121, the functional interrogation of mutations between host strains identified a single point mutation in the dltB gene. Such mutation was sufficient to cause the rabbit disease phenotype in animal models116; however, the mechanisms by which dltB mutations result in this host adaptation are yet to be defined (Fig. 4 and Supplementary Fig. 3).
The ability of S. aureus to adapt to new hosts, and the capacity to adapt to population bottlenecks, has been further explored in a model of human-to-ovine host switching complemented by large-scale genomic analyses117. This revealed that S. aureus could overcome substantial population bottlenecks and acquire beneficial mutations over time. However, there was not a strong signal of convergence, which suggests that multiple pathways to adaptation were occurring in this model. The exploitation of host-switching models will further enhance the understanding of adaptive mechanisms underlying species-specific pathogenesis.
Conclusions and outlook
S. aureus remains a formidable multispecies pathogen and major challenge to human health. Fuelled by the major progress in omics, important advances have been made in the past 10 years to understand the duality of S. aureus as an asymptomatic colonizer and devastating invasive pathogen. Although new immune evasion molecules and novel ligands for well-described molecules have been identified, a holistic understanding of the in vivo dynamics and interactions within host will be unlikely revealed by a traditional one-by-one gene discovery approach. The variability in the immune evasion repertoire across S. aureus clones and the heterogeneity of host responses substantially complicate this issue, particularly in complex clinical contexts. Thus, systems-level experimental approaches that account for S. aureus strain variation, combined with improved laboratory infection models (perhaps even controlled human infection or colonization models) that consider immune response and microbiome context, are needed to better understand the roles of critical S. aureus factors during infection. Such advances could help address the issues that have challenged S. aureus vaccine development such as immune imprinting resulting from previous S. aureus exposure.
With its capacity to switch between host species, transition from colonization to invasion, or develo** antibiotic resistance and persistence, S. aureus has the propensity to adapt to new environments. An integrated understanding of how S. aureus genomics, evolution and adaptation intersect with virulence and immune evasion strategies will guide the design of prevention strategies and therapeutic approaches. Other potential avenues could stem from the identification of human genetic signatures associated with S. aureus disease risk as well as from the microbiome composition of the skin, the nose and the gut.
It is also becoming increasingly apparent that immunometabolism has a key role in the pathogenesis of S. aureus infections. However, microbial metabolism has often been neglected, with many research groups instead using pathogen-associated molecular patterns as a proxy for bacterial stimulation. There is increasing evidence that staphylococcal metabolic flexibility is not only critical for bacterial persistence during infection but also in sha** the host immunometabolic response. Importantly, this metabolic interplay between host and S. aureus varies by infection site. Targeting immunometabolism as an alternative or complementary therapeutic strategy to enhance multidrug-resistant S. aureus clearance will become more realistic as our understanding of host–pathogen metabolic interactions deepens.
Ultimately, advancing the study of S. aureus host–pathogen interactions by acknowledging the roles of other microbiota and immunometabolism, and further understanding pathogen genetic variability and adaptation will generate holistic insights that lead to meaningful clinical impact on diseases caused by S. aureus.
References
Lowy, F. D. Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 (1998).
Wertheim, H. F. et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5, 751–762 (2005).
Williams, R. E. Healthy carriage of Staphylococcus aureus: its prevalence and importance. Bacteriol. Rev. 27, 56–71 (1963).
Krismer, B., Weidenmaier, C., Zipperer, A. & Peschel, A. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat. Rev. Microbiol. 15, 675–687 (2017). Excellent review on the nasal microbiome and the interaction with S. aureus.
Clarridge, J. E. III, Harrington, A. T., Roberts, M. C., Soge, O. O. & Maquelin, K. Impact of strain ty** methods on assessment of relationship between paired nares and wound isolates of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 51, 224–231 (2013).
Raineri, E. J. M., Altulea, D. & van Dijl, J. M. Staphylococcal trafficking and infection-from ‘nose to gut’ and back. FEMS Microbiol. Rev. 46, fuab041 (2022).
Spaan, A. N., van Strijp, J. A. G. & Torres, V. J. Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 15, 435–447 (2017). Comprehensive review of the staphylococcal leukocidins.
Thammavongsa, V., Kim, H. K., Missiakas, D. & Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 13, 529–543 (2015).
Montgomery, C. P., David, M. Z. & Daum, R. S. Host factors that contribute to recurrent staphylococcal skin infection. Curr. Opin. Infect. Dis. 28, 253–258 (2015).
Kallen, A. J. et al. Health care-associated invasive MRSA infections, 2005–2008. JAMA 304, 641–648 (2010).
Jansen, K. U., Girgenti, D. Q., Scully, I. L. & Anderson, A. S. Vaccine review: ‘Staphyloccocus aureus vaccines: problems and prospects’. Vaccine 31, 2723–2730 (2013).
Wollenberg, M. S. et al. Propionibacterium-produced coproporphyrin III induces Staphylococcus aureus aggregation and biofilm formation. mBio 5, e01286-14 (2014).
Costello, E. K. et al. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009).
Torres Salazar, B. O., Heilbronner, S., Peschel, A. & Krismer, B. Secondary metabolites governing microbiome interaction of Staphylococcal pathogens and commensals. Micro. Physiol. 31, 198–216 (2021).
Lister, J. L. & Horswill, A. R. Staphylococcus aureus biofilms: recent developments in biofilm dispersal. Front. Cell Infect. Microbiol. 4, 178 (2014).
Archer, N. K. et al. Staphylococcus aureus biofilms: properties, regulation, and roles in human disease. Virulence 2, 445–459 (2011).
Bhattacharya, M. et al. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc. Natl Acad. Sci. USA 115, 7416–7421 (2018).
Chambers, H. F. & Deleo, F. R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7, 629–641 (2009).
Turner, N. A. et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat. Rev. Microbiol. 17, 203–218 (2019).
Howden, B. P., Davies, J. K., Johnson, P. D., Stinear, T. P. & Grayson, M. L. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23, 99–139 (2010).
Gao, W. et al. The RpoB H481Y rifampicin resistance mutation and an active stringent response reduce virulence and increase resistance to innate immune responses in Staphylococcus aureus. J. Infect. Dis. 207, 929–939 (2013).
Proctor, R. A. et al. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4, 295–305 (2006).
Moller, A. G., Petit, R. A. III & Read, T. D. Species-scale genomic analysis of Staphylococcus aureus genes influencing phage host range and their relationships to virulence and antibiotic resistance genes. mSystems 7, e0108321 (2022).
Howden, B. P. et al. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 7, e1002359 (2011).
Mwangi, M. M. et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl Acad. Sci. USA 104, 9451–9456 (2007).
Gao, W. et al. Large tandem chromosome expansions facilitate niche adaptation during persistent infection with drug-resistant Staphylococcus aureus. Microb. Genom. 1, e000026 (2015).
Panton, P. N. & Valentine, F. C. O. Staphylococcal toxin. Lancet 219, 506–508 (1932).
Cheung, G. Y. C., Bae, J. S. & Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 12, 547–569 (2021).
Curnutte, J. T., Whitten, D. M. & Babior, B. M. Defective superoxide production by granulocytes from patients with chronic granulomatous disease. N. Engl. J. Med. 290, 593–597 (1974).
Spaan, A. N., Surewaard, B. G., Nijland, R. & van Strijp, J. A. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu. Rev. Microbiol. 67, 629–650 (2013).
Stapels, D. A. et al. Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc. Natl Acad. Sci. USA 111, 13187–13192 (2014).
Kretschmer, D. et al. Staphylococcus aureus depends on eap proteins for preventing degradation of its phenol-soluble modulin toxins by neutrophil serine proteases. Front. Immunol. 12, 701093 (2021).
Stapels, D. A. et al. Staphylococcus aureus protects its immune-evasion proteins against degradation by neutrophil serine proteases. Cell Microbiol. 18, 536–545 (2016).
Ploscariu, N. T., de Jong, N. W. M., van Kessel, K. P. M., van Strijp, J. A. G. & Geisbrecht, B. V. Identification and structural characterization of a novel myeloperoxidase inhibitor from Staphylococcus delphini. Arch. Biochem. Biophys. 645, 1–11 (2018).
de Jong, N. W. M. et al. Immune evasion by a staphylococcal inhibitor of myeloperoxidase. Proc. Natl Acad. Sci. USA 114, 9439–9444 (2017).
Loffler, B. et al. Staphylococcus aureus Panton-Valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PloS Pathog. 6, e1000715 (2010).
Vrieling, M. et al. Population analysis of Staphylococcus aureus reveals a cryptic, highly prevalent superantigen SelW that contributes to the pathogenesis of bacteremia. mBio 11, e02082-20 (2020).
Wilson, G. J. et al. A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia. PloS Pathog. 7, e1002271 (2011).
Tuffs, S. W. et al. The Staphylococcus aureus superantigen SelX is a bifunctional toxin that inhibits neutrophil function. PloS Pathog. 13, e1006461 (2017).
Foster, T. J., Geoghegan, J. A., Ganesh, V. K. & Hook, M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 12, 49–62 (2014).
Corrigan, R. M., Miajlovic, H. & Foster, T. J. Surface proteins that promote adherence of Staphylococcus aureus to human desquamated nasal epithelial cells. BMC Microbiol. 9, 22 (2009).
Cheng, A. G. et al. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23, 3393–3404 (2009).
Askarian, F. et al. Serine-aspartate repeat protein D increases Staphylococcus aureus virulence and survival in blood. Infect. Immun. 85, e00559-16 (2017).
Zhang, Y. et al. Staphylococcus aureus SdrE captures complement factor H’s C-terminus via a novel ‘close, dock, lock and latch’ mechanism for complement evasion. Biochem. J. 474, 1619–1631 (2017).
Speziale, P. & Pietrocola, G. The multivalent role of fibronectin-binding proteins A and B (FnBPA and FnBPB) of Staphylococcus aureus in host infections. Front. Microbiol. 11, 2054 (2020).
Pietrocola, G. et al. Fibronectin-binding protein B (FnBPB) from Staphylococcus aureus protects against the antimicrobial activity of histones. J. Biol. Chem. 294, 3588–3602 (2019).
Thammavongsa, V., Missiakas, D. M. & Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 342, 863–866 (2013).
Soh, K. Y., Loh, J. M. S. & Proft, T. Cell wall-anchored 5ʹ-nucleotidases in Gram-positive cocci. Mol. Microbiol. 113, 691–698 (2020).
Berends, E. T. M. et al. Staphylococcus aureus impairs the function of and kills human dendritic cells via the LukAB toxin. mBio 10, e01918-18 (2019).
Badarau, A., Trstenjak, N. & Nagy, E. Structure and function of the two-component cytotoxins of Staphylococcus aureus — learnings for designing novel therapeutics. Adv. Exp. Med. Biol. 966, 15–35 (2017).
Perelman, S. S. et al. Genetic variation of staphylococcal LukAB toxin determines receptor tropism. Nat. Microbiol. 6, 731–745 (2021).
Spaan, A. N. et al. The staphylococcal toxin Panton-Valentine leukocidin targets human C5a receptors. Cell Host Microbe 13, 584–594 (2013).
Haapasalo, K. et al. Staphylococcus aureus toxin LukSF dissociates from its membrane receptor target to enable renewed ligand sequestration. FASEB J. 33, 3807–3824 (2019).
Tromp, A. T. et al. Human CD45 is an F-component-specific receptor for the staphylococcal toxin Panton-Valentine leukocidin. Nat. Microbiol. 3, 708–717 (2018).
Knop, J. et al. Staphylococcus aureus infection in humanized mice: a new model to study pathogenicity associated with human immune response. J. Infect. Dis. 212, 435–444 (2015).
Prince, A., Wang, H., Kitur, K. & Parker, D. Humanized mice exhibit increased susceptibility to Staphylococcus aureus pneumonia. J. Infect. Dis. 215, 1386–1395 (2017).
Tseng, C. W. et al. Increased susceptibility of humanized NSG mice to Panton-Valentine leukocidin and Staphylococcus aureus skin infection. PLoS Pathog. 11, e1005292 (2015).
Muthukrishnan, G. et al. Humanized mice exhibit exacerbated abscess formation and osteolysis during the establishment of implant-associated Staphylococcus aureus osteomyelitis. Front. Immunol. 12, 651515 (2021).
Chow, S. H. et al. Targeting NLRP3 and staphylococcal pore-forming toxin receptors in human-induced pluripotent stem cell-derived macrophages. J. Leukoc. Biol. 108, 967–981 (2020).
Lees, J. A. & Bentley, S. D. Bacterial GWAS: not just gilding the lily. Nat. Rev. Microbiol. 14, 406 (2016).
Young, B. C. et al. Panton-Valentine leucocidin is the key determinant of Staphylococcus aureus pyomyositis in a bacterial GWAS. eLife 8, e42486 (2019). Statistical genomics study linking a S. aureus virulence factor to a clinical manifestation.
Koymans, K. J. et al. Staphylococcal superantigen-like protein 1 and 5 (SSL1 & SSL5) limit neutrophil chemotaxis and migration through MMP-inhibition. Int. J. Mol. Sci. 17, 1072 (2016).
Bestebroer, J. et al. Staphylococcal SSL5 inhibits leukocyte activation by chemokines and anaphylatoxins. Blood 113, 328–337 (2009).
Tang, A. et al. Staphylococcus aureus superantigen-like protein SSL1: a toxic protease. Pathogens 8, 2 (2019).
Koymans, K. J. et al. The TLR2 antagonist Staphylococcal superantigen-like protein 3 acts as a virulence factor to promote bacterial pathogenicity in vivo. J. Innate Immun. 9, 561–573 (2017).
Zhao, Y. et al. Staphylococcal superantigen-like protein 13 activates neutrophils via formyl peptide receptor 2. Cell Microbiol. 20, e12941 (2018).
Diebolder, C. A. et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 343, 1260–1263 (2014).
Becker, S., Frankel, M. B., Schneewind, O. & Missiakas, D. Release of protein A from the cell wall of Staphylococcus aureus. Proc. Natl Acad. Sci. USA 111, 1574–1579 (2014).
O’Halloran, D. P., Wynne, K. & Geoghegan, J. A. Protein A is released into the Staphylococcus aureus culture supernatant with an unprocessed sorting signal. Infect. Immun. 83, 1598–1609 (2015).
Falugi, F., Kim, H. K., Missiakas, D. M. & Schneewind, O. Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus. mBio 4, e00575-13 (2013).
Cruz, A. R. et al. Staphylococcal protein A inhibits complement activation by interfering with IgG hexamer formation. Proc. Natl Acad. Sci. USA 118, e2016772118 (2021).
Hong, X. et al. Staphylococcal protein A promotes colonization and immune evasion of the epidemic healthcare-associated MRSA ST239. Front. Microbiol. 7, 951 (2016).
Smith, E. J. et al. The immune evasion protein Sbi of Staphylococcus aureus occurs both extracellularly and anchored to the cell envelope by binding lipoteichoic acid. Mol. Microbiol. 83, 789–804 (2012).
Dunphy, R. W. et al. Staphylococcal complement evasion protein Sbi stabilises C3d dimers by inducing an N-terminal helix swap. Front. Immunol. 13, 892234 (2022).
Dasari, P. et al. The protease SplB of Staphylococcus aureus targets host complement components and inhibits complement-mediated bacterial opsonophagocytosis. J. Bacteriol. 204, e0018421 (2022).
Bhattacharya, M. et al. Leukocidins and the nuclease nuc prevent neutrophil-mediated killing of Staphylococcus aureus biofilms. Infect. Immun. 88, e00372-20 (2020).
Schilcher, K. & Horswill, A. R. Staphylococcal biofilm development: structure, regulation, and treatment strategies. Microbiol. Mol. Biol. Rev. 84, e00026-19 (2020). Comprehensive review of S. aureus biofilms.
Ricciardi, B. F. et al. Staphylococcus aureus evasion of host immunity in the setting of prosthetic joint infection: biofilm and beyond. Curr. Rev. Musculoskelet. Med. 11, 389–400 (2018).
Arciola, C. R., Campoccia, D. & Montanaro, L. Implant infections: adhesion, biofilm formation and immune evasion. Nat. Rev. Microbiol. 16, 397–409 (2018).
Heim, C. E., Vidlak, D. & Kielian, T. Interleukin-10 production by myeloid-derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J. Leukoc. Biol. 98, 1003–1013 (2015).
Heim, C. E. et al. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J. Immunol. 194, 3861–3872 (2015).
Heim, C. E. et al. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J. Immunol. 192, 3778–3792 (2014).
Heim, C. E., West, S. C., Ali, H. & Kielian, T. Heterogeneity of Ly6G(+) Ly6C(+) myeloid-derived suppressor cell infiltrates during Staphylococcus aureus biofilm infection. Infect. Immun. 86, e00684-18 (2018).
Yamada, K. J. et al. Monocyte metabolic reprogramming promotes pro-inflammatory activity and Staphylococcus aureus biofilm clearance. PLoS Pathog. 16, e1008354 (2020).
Gabrilovich, D. I. & Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 (2009).
O’Neill, L. A., Kishton, R. J. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013).
Lampropoulou, V. et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166 (2016).
Hooftman, A. et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. 32, 468–478.e7 (2020).
Qin, W. et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol. 15, 983–991 (2019).
Heim, C. E. et al. Lactate production by Staphylococcus aureus biofilm inhibits HDAC11 to reprogramme the host immune response during persistent infection. Nat. Microbiol. 5, 1271–1284 (2020). Sophisticated host–pathogen study using at-scale transposon mutagenesis to reveal the role mechanism of S. aureus lactate in regulation of IL-10 expression.
Tomlinson, K. L. & Riquelme, S. A. Host–bacteria metabolic crosstalk drives S. aureus biofilm. Microb. Cell 8, 106–107 (2021).
Tomlinson, K. L. et al. Staphylococcus aureus induces an itaconate-dominated immunometabolic response that drives biofilm formation. Nat. Commun. 12, 1399 (2021).
Michelucci, A. et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl Acad. Sci. USA 110, 7820–7825 (2013).
Arts, R. J. et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 24, 807–819 (2016).
Chan, L. C. et al. Innate immune memory contributes to host defense against recurrent skin and skin structure infections caused by methicillin-resistant Staphylococcus aureus. Infect. Immun. 85, e00876-16 (2017).
Chan, L. C. et al. Protective immunity in recurrent Staphylococcus aureus infection reflects localized immune signatures and macrophage-conferred memory. Proc. Natl Acad. Sci. USA 115, E11111–E11119 (2018).
Acker, K. P. et al. Strains of Staphylococcus aureus that colonize and infect skin harbor mutations in metabolic genes. iScience 19, 281–290 (2019).
Wong Fok Lung, T. et al. Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat. Microbiol. 5, 141–153 (2020).
Proctor, R. A., van Langevelde, P., Kristjansson, M., Maslow, J. N. & Arbeit, R. D. Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin. Infect. Dis. 20, 95–102 (1995).
Kriegeskorte, A. et al. Staphylococcus aureus small colony variants show common metabolic features in central metabolism irrespective of the underlying auxotrophism. Front. Cell Infect. Microbiol. 4, 141 (2014).
Gabryszewski, S. J. et al. Metabolic adaptation in methicillin-resistant Staphylococcus aureus pneumonia. Am. J. Respir. Cell Mol. Biol. 61, 185–197 (2019).
Guerillot, R. et al. Unstable chromosome rearrangements in Staphylococcus aureus cause phenotype switching associated with persistent infections. Proc. Natl Acad. Sci. USA 116, 20135–20140 (2019). Identification of a reversible genome rearrangement contributing to the SCV phenotype in S. aureus.
Richardson, E. J. et al. Gene exchange drives the ecological success of a multi-host bacterial pathogen. Nat. Ecol. Evol. 2, 1468–1478 (2018). Excellent demonstration of bacterial population genomics to reveal the extent to which humans are sha** the evolution of S. aureus and exchange between different animal hosts.
Price, L. B. et al. Staphylococcus aureus CC398: host adaptation and emergence of methicillin resistance in livestock. mBio 3, e00305-11 (2012).
Hau, S. J., Sun, J., Davies, P. R., Frana, T. S. & Nicholson, T. L. Comparative prevalence of immune evasion complex genes associated with beta-hemolysin converting bacteriophages in MRSA ST5 isolates from swine, swine facilities, humans with swine contact, and humans with no swine contact. PLoS ONE 10, e0142832 (2015).
Sahibzada, S. et al. Transmission of highly virulent community-associated MRSA ST93 and livestock-associated MRSA ST398 between humans and pigs in Australia. Sci. Rep. 7, 5273 (2017).
Spoor, L. E. et al. Livestock origin for a human pandemic clone of community-associated methicillin-resistant Staphylococcus aureus. mBio 4, e00356-13 (2013).
Larsen, J. et al. Emergence of methicillin resistance predates the clinical use of antibiotics. Nature 602, 135–141 (2022). Large-scale genomic study identifying an animal source for antimicrobial-resistant S. aureus in humans.
Hallatschek, O., Hersen, P., Ramanathan, S. & Nelson, D. R. Genetic drift at expanding frontiers promotes gene segregation. Proc. Natl Acad. Sci. USA 104, 19926–19930 (2007).
Nei, M. Selectionism and neutralism in molecular evolution. Mol. Biol. Evol. 22, 2318–2342 (2005).
McVicker, G. et al. Clonal expansion during Staphylococcus aureus infection dynamics reveals the effect of antibiotic intervention. PLoS Pathog. 10, e1003959 (2014).
Didelot, X., Walker, A. S., Peto, T. E., Crook, D. W. & Wilson, D. J. Within-host evolution of bacterial pathogens. Nat. Rev. Microbiol. 14, 150–162 (2016).
Sheppard, S. K., Guttman, D. S. & Fitzgerald, J. R. Population genomics of bacterial host adaptation. Nat. Rev. Genet. 19, 549–565 (2018). Comprehensive review of bacterial host species adaptation.
Guinane, C. M. et al. Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome Biol. Evol. 2, 454–466 (2010).
Viana, D. et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat. Genet. 47, 361–366 (2015).
Bacigalupe, R., Tormo-Mas, M. A., Penades, J. R. & Fitzgerald, J. R. A multihost bacterial pathogen overcomes continuous population bottlenecks to adapt to new host species. Sci. Adv. 5, eaax0063 (2019). Experimental study demonstrating the capacity of S. aureus to acquire beneficial mutations alleviating evolutionary bottlenecks and enabling its adaptation to different hosts.
Zhou, W. et al. WGS analysis of ST9-MRSA-XII isolates from live pigs in China provides insights into transmission among porcine, human and bovine hosts. J. Antimicrob. Chemother. 73, 2652–2661 (2018).
Haag, A. F., Fitzgerald, J. R. & Penades, J. R. Staphylococcus aureus in animals. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.GPP3-0060-2019 (2019).
Vrieling, M. et al. Bovine Staphylococcus aureus secretes the leukocidin LukMF’ to kill migrating neutrophils through CCR1. mBio 6, e00335 (2015).
Matuszewska, M., Murray, G. G. R., Harrison, E. M., Holmes, M. A. & Weinert, L. A. The evolutionary genomics of host specificity in Staphylococcus aureus. Trends Microbiol. 28, 465–477 (2020).
Park, S. & Ronholm, J. Staphylococcus aureus in agriculture: lessons in evolution from a multispecies pathogen. Clin. Microbiol. Rev. 34, e00182-20 (2021).
de Jong, N. W. M. et al. Identification of a staphylococcal complement inhibitor with broad host specificity in equid Staphylococcus aureus strains. J. Biol. Chem. 293, 4468–4477 (2018).
Petit, R. A. III & Read, T. D. Staphylococcus aureus viewed from the perspective of 40,000+ genomes. Peer J. 6, e5261 (2018).
Lindsay, J. A. Genomic variation and evolution of Staphylococcus aureus. Int. J. Med. Microbiol. 300, 98–103 (2010).
Malachowa, N. & DeLeo, F. R. Mobile genetic elements of Staphylococcus aureus. Cell Mol. Life Sci. 67, 3057–3071 (2010).
Golubchik, T. et al. Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS ONE 8, e61319 (2013).
Everitt, R. G. et al. Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus. Nat. Commun. 5, 3956 (2014).
Prunier, A.-L. et al. High rate of macrolide resistance in Staphylococcus aureus strains from patients with cystic fibrosis reveals high proportions of hypermutable strains. J. Infect. Dis. 187, 1709–1716 (2003).
Giulieri, S. G. et al. Niche-specific genome degradation and convergent evolution sha** Staphylococcus aureus adaptation during severe infections. eLife 11, e77195 (2022). Population genomics conducted on a very large clinical cohort to reveal with unprecedented resolution the bacterial genome-wide changes that are associated with the transition from colonizing to invasive S. aureus infections.
Giulieri, S. G. et al. Genomic exploration of sequential clinical isolates reveals a distinctive molecular signature of persistent Staphylococcus aureus bacteraemia. Genome Med. 10, 65 (2018).
Klemm, E. J. et al. Emergence of host-adapted Salmonella Enteritidis through rapid evolution in an immunocompromised host. Nat. Microbiol. 1, 15023 (2016).
Batut, B., Knibbe, C., Marais, G. & Daubin, V. Reductive genome evolution at both ends of the bacterial population size spectrum. Nat. Rev. Microbiol. 12, 841–850 (2014).
Hall, M. D. et al. Improved characterisation of MRSA transmission using within-host bacterial sequence diversity. eLife 8, 46402 (2019).
Long, D. R. et al. Polyclonality, shared strains, and convergent evolution in chronic CF S. aureus airway infection. Am. J. Respir. Crit. Care Med. 203, 1127–1137 (2021).
Young, B. C. et al. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc. Natl Acad. Sci. USA 109, 4550–4555 (2012).
Young, B. C. et al. Severe infections emerge from commensal bacteria by adaptive evolution. eLife 6, e30637 (2017).
Altman, D. R. et al. Genome plasticity of agr-defective Staphylococcus aureus during clinical infection. Infect. Immun. 86, e00331-18 (2018).
Das, S. et al. Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation. Proc. Natl Acad. Sci. USA 113, E3101–E3110 (2016).
Paudel, A., Panthee, S., Hamamoto, H., Grunert, T. & Sekimizu, K. YjbH regulates virulence genes expression and oxidative stress resistance in Staphylococcus aureus. Virulence 12, 470–480 (2021).
Jousselin, A., Kelley, W. L., Barras, C., Lew, D. P. & Renzoni, A. The Staphylococcus aureus Thiol/oxidative stress global regulator Spx controls trfA, a gene implicated in cell wall antibiotic resistance. Antimicrob. Agents Chemother. 57, 3283–3292 (2013).
Lopatkin, A. J. et al. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 371, eaba0862 (2021).
Elgrail, M. M. et al. Convergent evolution of antibiotic tolerance in patients with persistent methicillin-resistant Staphylococcus aureus bacteremia. Infect. Immun. 90, e0000122 (2022).
Giulieri, S. G. et al. Comprehensive genomic investigation of adaptive mutations driving the low-level oxacillin resistance phenotype in Staphylococcus aureus. mBio 11, e02882-20 (2020).
Howden, B. P., Johnson, P. D., Ward, P. B., Stinear, T. P. & Davies, J. K. Isolates with low-level vancomycin resistance associated with persistent methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 50, 3039–3047 (2006).
Loss, G. et al. Staphylococcus aureus small colony variants (SCVs): news from a chronic prosthetic joint infection. Front. Cell Infect. Microbiol. 9, 363 (2019).
Bär, J. et al. Quantification of within-patient Staphylococcus aureus phenotypic heterogeneity as a proxy for presence of persisters across clinical presentations. Clin. Microbiol. Infect. 28, 1022.e1–1022.e7 (2022).
Laabei, M. et al. Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus. PLoS Biol. 13, e1002229 (2015).
**ong, Y. Q. et al. Phenotypic and genotypic characteristics of persistent methicillin-resistant Staphylococcus aureus bacteremia in vitro and in an experimental endocarditis model. J. Infect. Dis. 199, 201–208 (2009).
Scott, W. K. et al. Human genetic variation in GLS2 is associated with development of complicated Staphylococcus aureus bacteremia. PLoS Genet. 14, e1007667 (2018).
Spaan, A. N. et al. Human OTULIN haploinsufficiency impairs cell-intrinsic immunity to staphylococcal alpha-toxin. Science 376, eabm6380 (2022). This is the first evidence of a human heterozygous gene deficiency predisposing patients to severe S. aureus infection.
Chang, Y. L. et al. Human DNA methylation signatures differentiate persistent from resolving MRSA bacteremia. Proc. Natl Acad. Sci. USA 118, e2000663118 (2021).
Ford, C. A., Hurford, I. M. & Cassat, J. E. Antivirulence strategies for the treatment of Staphylococcus aureus infections: a mini review. Front. Microbiol. 11, 632706 (2020).
Francois, B. et al. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: first-in-human trial. Intensive Care Med. 44, 1787–1796 (2018).
Magyarics, Z. et al. Randomized, double-blind, placebo-controlled, single-ascending-dose study of the penetration of a monoclonal antibody combination (ASN100) targeting Staphylococcus aureus cytotoxins in the lung epithelial lining fluid of healthy volunteers. Antimicrob. Agents Chemother. 63, e00350-19 (2019).
Chan, R. et al. Identification of biologic agents to neutralize the bicomponent leukocidins of Staphylococcus aureus. Sci. Transl Med. 11, eaat0882 (2019).
Mansson, M. et al. Inhibition of virulence gene expression in Staphylococcus aureus by novel depsipeptides from a marine photobacterium. Mar. Drugs 9, 2537–2552 (2011).
Gao, P., Davies, J. & Kao, R. Y. T. Dehydrosqualene desaturase as a novel target for anti-virulence therapy against Staphylococcus aureus. mBio 8, e01224-17 (2017).
Chen, X., Schneewind, O. & Missiakas, D. Engineered human antibodies for the opsonization and killing of Staphylococcus aureus. Proc. Natl Acad. Sci. USA 119, e2114478119 (2022).
Miller, L. S., Fowler, V. G., Shukla, S. K., Rose, W. E. & Proctor, R. A. Development of a vaccine against Staphylococcus aureus invasive infections: evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol. Rev. 44, 123–153 (2020).
Tsai, C. M. et al. Non-protective immune imprint underlies failure of Staphylococcus aureus IsdB vaccine. Cell Host Microbe 30, 1163–1172 (2022).
Nakatsuji, T. et al. Development of a human skin commensal microbe for bacteriotherapy of atopic dermatitis and use in a phase 1 randomized clinical trial. Nat. Med. 27, 700–709 (2021). Clinical study of bacteriotherapy highlighting that the inhibition of PSM production by commensal S. hominis decreases S. aureus colonization in atopic dermatitis patients.
Liu, Y. et al. Skin microbiota analysis-inspired development of novel anti-infectives. Microbiome 8, 85 (2020).
Piewngam, P. & Otto, M. Probiotics to prevent Staphylococcus aureus disease? Gut Microbes 11, 94–101 (2020).
Olagnier, D. et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 11, 4938 (2020).
Horn, C. M. & Kielian, T. Crosstalk between Staphylococcus aureus and innate immunity: focus on immunometabolism. Front. Immunol. 11, 621750 (2020).
Prince, A. & Wong Fok Lung, T. Consequences of metabolic interactions during Staphylococcus aureus infection. Toxins 12, 581 (2020).
Thurlow, L. R. et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 186, 6585–6596 (2011).
Pleasance, E. et al. Whole genome and transcriptome analysis enhances precision cancer treatment options. Ann. Oncol. 33, 939–949 (2022).
Recker, M. et al. Clonal differences in Staphylococcus aureus bacteraemia-associated mortality. Nat. Microbiol. 2, 1381–1388 (2017).
Young, B. C. et al. Antimicrobial resistance determinants are associated with Staphylococcus aureus bacteraemia and adaptation to the healthcare environment: a bacterial genome-wide association study. Microb. Genom. 7, 000700 (2021).
Lilje, B. et al. Whole-genome sequencing of bloodstream Staphylococcus aureus isolates does not distinguish bacteraemia from endocarditis. Microb. Genom. https://doi.org/10.1099/mgen.0.000138 (2017).
Denamur, E. et al. Genome wide association study of Escherichia coli bloodstream infection isolates identifies genetic determinants for the portal of entry but not fatal outcome. PLoS Genet. 18, e1010112 (2022).
Wozniak, J. M. et al. Mortality risk profiling of Staphylococcus aureus bacteremia by multi-omic serum analysis reveals early predictive and pathogenic signatures. Cell 182, 1311–1327 (2020).
Acknowledgements
This work was supported by the National Health and Medical Research Council Australia through Investigator Grants to B.P.H. (GNT1196103) and T.P.S. (GNT1194325).
Author information
Authors and Affiliations
Contributions
B.P.H., T.P.S., S.G.G., S.L.B., L.K.S., J.Y.H.L., A.H., I.R.M. and T.W.F.L. researched data for the article. B.P.H., T.P.S., S.G.G., S.L.B., J.Y.H.L., A.H., I.R.M. and T.W.F.L. substantially contributed to discussion of content. B.P.H. and T.P.S. led the writing of the article with contributions from S.G.G., J.Y.H.L., A.H., I.R.M. and T.W.F.L. B.P.H., T.P.S., S.G.G., L.K.S., A.H., I.R.M. and T.W.F.L. reviewed and edited the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Microbiology thanks Jos van Strijp, Timothy Foster and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Accessory genome
-
Genes usually associated with mobile genetic elements that are present in only a subset of S. aureus strains. The accessory genome is one cause of variability in strain behaviour.
- Biofilms
-
A sessile microbial community usually enclosed by a protective extracellular matrix and attached to a surface or other cells.
- Bottlenecks
-
When a population (for example, bacterial population) is significantly reduced in size, limiting genetic diversity.
- Carbon catabolite repression
-
(CCR). A bacterial global regulatory process that results in the selective use of substrates from a mixture of carbon sources.
- Chronic granulomatous diseases
-
Rare X-linked recessive inherited immune deficiencies caused by defects in the enzyme, NADPH oxidase resulting in phagocytic dysfunction.
- Colonization
-
The presence of S. aureus on a body site such as the skin, gut or anterior nares, without causing disease.
- Core genome
-
Represents genes that are present in all S. aureus strains.
- Efferocytosis
-
A process of phagocytic engulfment of dead or dying cells.
- Evasion
-
Strategies used by bacteria to evade killing by the immune system.
- Genetic drift
-
A change in the frequency of an existing genetic variant in a population owing to random chance.
- Genome-wide association studies
-
The use of statistical genomics methods to identify the genetic variants linked to a particular phenotype.
- Insertion sequences
-
A short segment of DNA that can move within the S. aureus chromosome as a simple transposable element and contribute to bacterial adaptation.
- Invasion
-
The ability of a bacterial pathogen to spread to other locations in the host by invading host cells, such as the transition from the anterior nares (colonization) to the bloodstream (invasion).
- Leukotoxins
-
Toxin proteins that penetrate lipid bilayers to form pores.
- Lipopolysaccharide
-
Important outer membrane component of Gram-negative bacterial cell walls that acts as an endotoxin.
- Machine learning
-
The application of computer systems using statistical models and algorithms to draw conclusions from data.
- Microbial surface components recognizing adhesive matrix molecules
-
(MSCRAMMs). Adhesin proteins that are important in the initial binding of S. aureus to host tissues.
- Mobile genetic elements
-
Sequences of genetic material that can change places in the S. aureus chromosome or move between bacterial chromosomes.
- Persistence
-
Broadly refers to the ability of bacterial cells, including S. aureus, to cause persistent infection, despite the activity of the immune system or antibiotic therapy.
- Pyomyositis
-
Deep infection in the skeletal muscles, usually associated with abscess formation.
- Recurrent
-
The propensity for S. aureus infection to recur after initial successful therapy through surgery and/or antibiotics.
- Small colony variant
-
(SCV). A slow-growing (small colony) population of S. aureus that is associated with persistent and recurrent infections.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Howden, B.P., Giulieri, S.G., Wong Fok Lung, T. et al. Staphylococcus aureus host interactions and adaptation. Nat Rev Microbiol 21, 380–395 (2023). https://doi.org/10.1038/s41579-023-00852-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-023-00852-y
- Springer Nature Limited
This article is cited by
-
Development and characterization of a topical gel, containing lavender (Lavandula angustifolia) oil loaded solid lipid nanoparticles
BMC Complementary Medicine and Therapies (2024)
-
Demographic fluctuations in bloodstream Staphylococcus aureus lineages configure the mobile gene pool and antimicrobial resistance
npj Antimicrobials and Resistance (2024)
-
A guide to complement biology, pathology and therapeutic opportunity
Nature Reviews Immunology (2024)
-
Multimodal nanoimmunotherapy engages neutrophils to eliminate Staphylococcus aureus infections
Nature Nanotechnology (2024)
-
A Method Based on a Modified Fluorescence In Situ Hybridization (FISH) Approach for the Sensing of Staphylococcus aureus from Nasal Samples
Applied Biochemistry and Biotechnology (2024)