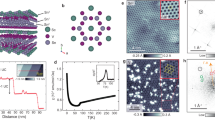
Basic characterizations of ScV6Sn6
We start with the basic characterizations of ScV6Sn6. Our transport measurements revealed a sudden change in electrical resistivity around TCO ≈ 92 K, signaling a symmetry-breaking phase transition (Fig. 1f). X-ray diffraction measurements detected commensurate superlattice peaks in the low-temperature phase consistent with the \(\sqrt{3}\)×\(\sqrt{3}\) charge ordering (Fig. 1d). Both the abrupt drop in the diffraction peak intensity at TCO (Fig. 1e) and the small thermal hysteresis in resistivity (Fig. 1f) are indicative of the first-order nature of the transition. Overall, our transport and diffraction characterizations of ScV6Sn6 are in close agreement with the original report37.
Before discussing the detailed electronic structure of ScV6Sn6, we briefly remark on the possible surface terminations of the 166 kagome materials. As shown in Fig. 1c, the unit cell of ScV6Sn6 consists of one ScSn2 layer, one hexagonal Sn2 layer, and two V3Sn kagome layers; this HfFe6Ge6-type 166 structure can expose complex surface terminations upon cleaving. We note that previous studies on the 166 kagome materials yield inconsistent interpretations on the surface terminations (Supplementary Note 1). To resolve this issue, we performed spatially resolved ARPES and XPS experiments on ScV6Sn6 using micro-focused synchrotron radiation (Fig. 1k, l). As summarized in Fig. 1m–o, we clearly identified three different surface domains characterized by dramatically different valence band structures and Sn 4d3/2, 4d5/2 core level spectra (D1, D2, and D3 domains, respectively). By comparing the ARPES and XPS spectra at each domain to the slab DFT calculations of various geometries, we unambiguously assigned the D1, D2, and D3 domains to the ScSn2, V3Sn, and Sn2 surface terminations, respectively (Supplementary Note 2, 3). Below we focus on the results obtained on the V3Sn termination (D2), which best represents the bulk electronic structure of ScV6Sn6 based on the slab calculation (Supplementary Note 3).
van Hove singularities in ScV6Sn6
Figure 2 displays our analysis of the low-energy electronic structure of ScV6Sn6. Similar to the case of AV3Sb5, we identified multiple kagome-derived vHSs near the Fermi level. In Fig. 2a–c, we present a three-dimensional stack of the ARPES spectra measured at the vicinity of M point (see the momentum positions of the cut 1–4 in Fig. 2j, see also Supplementary Fig. 4 for the ARPES spectra in the extended momentum range). From these plots, one can comprehensively understand the dispersions along both the Γ-M-Γ (see solid lines in the cut 4) and the K-M-K direction (see dashed lines across the cut 1 to 4). As shown in Fig. 2a, b, we identified two bands having electron-like character along the K-M-K direction and hole-like character along the Γ-M-Γ direction; these bands thus form saddle point structures or vHSs at the M point as predicted from the model kagome lattice dispersion (see also schematics in Fig. 2g, h). As shown in Fig. 2c, we also observed one additional vHS with inverted concavity, i.e., hole-like dispersion along the K-M-K and electron-like dispersion along the Γ-M-Γ direction (see schematics in Fig. 2i). The density functional theory (DFT) calculations in Fig. 2d–f closely reproduce the experimental results, revealing that the three vHSs in Fig. 2a–c respectively originate from the dxy, dxz, and dz2 local orbital degrees of freedom in the V kagome lattice (Supplementary Note 4). In the kagome lattice, the sublattice character of vHS – pure (p) or mixed (m) sublattice character – is also a topic of great interest, which critically determines the relevance of the on-site and long-range Coulomb interactions and the leading electronic instabilities3,4,23. By analyzing the sublattice weight distribution near the M point, we revealed that all three vHSs in ScV6Sn6 are p-type vHs having pure sublattice character (Supplementary Note 5). In sum, our analysis provides the complete characterizations of the dispersions, orbital characters, and sublattice types of the vHSs in ScV6Sn6.
Notably, the dxy and dz2 vHS of ScV6Sn6 locate very close to the Fermi level at –0.02 ± 0.01 eV and –0.03 ± 0.02 (Fig. 2a, c), while the dxz vHS is positioned at higher binding energy ≈ –0.40 ± 0.03 eV (Fig. 2b). The former vHSs contribute to the diverging density of states at the Fermi level and can in principle promote various electronic instabilities including the charge orderings. This scenario indeed applies to the case of AV3Sb5, where the vHSs at the Fermi level develop charge order gaps and directly contribute to the stabilization of the 2 × 2 charge order21,22,38,39. To test this scenario in ScV6Sn6, we tracked the temperature evolution of the vHSs across the \(\sqrt{3}\)×\(\sqrt{3}\) charge order transition. Comparing the ARPES dispersions in the normal (Fig. 2k, m) and charge-ordered states (Fig. 2l, n), we observed that all vHSs in ScV6Sn6 stay surprisingly unaltered across TCO, despite the fact that the vHS momentum (i.e., M point) lies at the folded Brillouin zone boundary of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order (see the schematics in Fig. 2j). This observation indicates that in stark contrast to the case of AV3Sb5, the intrinsic electronic instability of the V kagome lattice plays a marginal role in driving the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in ScV6Sn6.
Electronic reconstruction and charge order gap
After ruling out the vHS, we explore the electronic structure of ScV6Sn6 in the extended momentum range (Fig. 3), to identify the bands actually relevant to the \(\sqrt{3}\)×\(\sqrt{3}\) charge order transition. Figure 3a, b displays the Fermi surfaces of ScV6Sn6 measured at the normal and charge-ordered state, respectively. The major reconstruction of the Fermi surface across TCO is apparent from our data: the circular intensity pattern centered at \(\bar{\Gamma }\) in the normal state is modified to the star-shaped pattern in the charge-ordered state as highlighted with the cyan and orange guidelines. To better understand this change, we also present the corresponding energy-momentum dispersions along the \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) direction in Fig. 3e, f. In the normal state dispersion (Fig. 3e), we observe a large electron pocket centered at \(\bar{\Gamma }\), which constructs the circular intensity pattern in the normal state Fermi surface (Fig. 3a). Below TCO (Fig. 3f), this electron band bends toward the higher binding energy and develops a substantial charge order gap at the Fermi level. This opening of the charge order gap depletes the intensity in the Fermi surface along the \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) direction and explains the star-shaped Fermi surface observed in the charge-ordered state (Fig. 3b). The momentum position of the charge order gap is at about two-thirds of the \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) direction, which excellently matches with the folded Brillouin zone boundary of the \(\sqrt{3}\)×\(\sqrt{3}\) phase (Fig. 2j). We note that the band renormalization and charge order gap is also observed in other surface terminations supporting their bulk origin (see Supplementary Fig. 8 for the charge order gaps measured in the D1 termination). As shown in Fig. 3g, h, our DFT calculations closely capture the experimental results, reproducing the large electron pocket at \(\bar{\Gamma }\) in the normal state (Fig. 3g) and opening of the charge order gap ΔCO ≈ 260 meV across the Fermi level in the charge order state (Fig. 3h). Notably, the magnitude of the charge order gap in ScV6Sn6 is significantly larger than the ΔCO ≈ 80 meV of AV3Sb5 despite the comparable TCO in two systems21,22.
Microscopic origin of charge order in ScV6Sn6
Importantly, this large electron pocket at \(\bar{\Gamma }\), which is closely tied to the \(\sqrt{3}\)×\(\sqrt{3}\) charge order, has dominant planar Sn character (i.e., Sn(1) in Fig. 1c). To illustrate this, we present the DFT band structure of ScV6Sn6 in Fig. 4a, along with the V and Sn(1) orbital-projected calculations in Fig. 4b, c. The corresponding Fermi surfaces are also shown in the insets. In the V orbital-projected calculation (Fig. 4b), multiple Dirac bands at \(\bar{{\rm{K}}}\) and van Hove singularities at \(\bar{{\rm{M}}}\) originating from the V kagome lattice can be clearly identified. Overall, the V spectral weights dominate the Fermi surface near the zone boundary. In contrast, the Fermi surface near the zone center \(\bar{\Gamma }\) has dominant Sn(1) orbital character, as shown in the inset of Fig. 4c. We emphasize that it is this Sn(1) band at \(\bar{\Gamma }\) that develops the charge order gap and reconstructs the Fermi surface across TCO (Fig. 3), while the V kagome bands near \(\bar{{\rm{K}}}\) and \(\bar{{\rm{M}}}\) remain unaltered across TCO (Fig. 2). Our results thus highlight that the \(\sqrt{3}\)×\(\sqrt{3}\) charge order of ScV6Sn6 is tied to the structural components other than the V kagome lattice, especially to the planar Sn atoms.
The above conclusion from the electronic sector is further supported by our phonon mode calculations presented in Fig. 4d–g. As shown in Fig. 4d, the phonon dispersions of ScV6Sn6 display the continuum of unstable phonon modes centered at H, consistent with the \(\sqrt{3}\)×\(\sqrt{3}\) reconstruction in ScV6Sn6 at low temperature. By projecting the phonon density of states to the Sc, V, and Sn(1), Sn(2), Sn(3) sites in the unit cell, we revealed that the unstable phonon modes are associated with the structural distortions involving the planar Sn(1) and Sc sites, while the contribution from the V kagome layer is negligible (Fig. 4e). This result not only explains our observation of the large charge order gap on the Sn(1) bands (Fig. 3) and the marginal change of the V kagome bands (Fig. 2), but also is fully consistent with the X-ray refined charge order structure of ScV6Sn6 that revealed the dominant distortions in the Sn(1) and Sc sites37. It is also instructive to compare the phonon modes of ScV6Sn6 to those of the CsV3Sb5 shown in Fig. 4f, g. In stark contrast to the case of ScV6Sn6, the unstable phonon modes of CsV3Sb5 at M and L (associated with the 2 × 2 charge order) accompany the dominant displacement of the V atoms, and reflect the intrinsic electronic instability from the V kagome layers (Supplementary Fig. 11)40.
Phase diagram of charge order in Sc(V1−xCrx)6Sn6
Finally, we construct the phase diagram of Sc(V1−xCrx)6Sn6 series to understand the evolution of charge order with carrier do** (Fig. 5). The charge order phase remains robust in the wide-do** range, up to do** x ≈ 0.10 charges per V atom. In the framework of the virtual crystal approximation (Supplementary Fig. 12), this indicates that the charge order phase remains stable up to the order of 120 meV Fermi level shift, further making the electronic scenarios sensitive to the Fermi level, such as the Fermi surface or vHS nesting, unlikely. We note that this response of the charge order to carrier do** is again highly different from CsV3Sb5, where the charge order rapidly vanishes after the x ≈ 0.02 ~ 0.03 charge do** per V atom, regardless of the do** methods31,41. Such a rapid suppression of charge order in CsV3Sb5 with a slight shift of the chemical potential (that disturbs the nesting conditions) is consistent with the electronic origin of the charge order in AV3Sb5. Meanwhile, our powder X-ray diffraction characterizations of the Sc(V1-xCrx)6Sn6 series revealed that Cr substitution mainly affects the c-axis lattice parameter (Supplementary Fig. 13). Given that the \(\sqrt{3}\)×\(\sqrt{3}\) order in ScV6Sn6 mainly accompanies the c-axis motion of the Sc and Sn(1) atoms37, we suspect that the reduction of the c-axis lattice parameter is the main driving force of the charge order suppression in the Sc(V1-xCrx)6Sn6 phase diagram.
In summary, the present work elucidates the origin of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in the newly discovered kagome metal ScV6Sn6. Our comprehensive characterizations of the electronic structure, phonon dispersion, and phase diagram coherently emphasize the essential role of the structural degrees of freedom other than the V kagome lattice in driving the charge order. In this context, the nature of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in ScV6Sn6 is fundamentally different from the 2×2 charge order in the archetype kagome metal AV3Sb5, where the electronic instability in the V kagome lattice plays a major role. As discussed in the introduction, the true charge disproportionation phases in the kagome lattice can support the exotica of physics, including the fractionalization of elementary particles. While ScV6Sn6 is not an ideal platform to serve this purpose, our study underscores that the search for new kagome quantum materials hosting various types of genuine charge orders should be continued.
Note added in proof: while preparing the manuscript, relevant works on the electronic structure and phonon dispersions of ScV6Sn6 were reported42,43,\(\sqrt{3}\)×\(\sqrt{3}\)×3 charge order, all displaying the same temperature-dependence.
ARPES experiments
ARPES experiments were conducted at Beamline 7.0.2 (MAESTRO) and Beamline 4.0.3 (MERLIN) of the Advanced Light Source, equipped with R4000 and R8000 hemispherical electron analysers (Scienta Omicron), respectively. The samples were cleaved inside ultra-high vacuum chambers with a base pressure better than 4 × 10−11 Torr. To identify high-symmetry planes of the three-dimensional bulk Brillouin zone of ScV6Sn6, the photon energy dependent ARPES measurements were performed in a wide phonon energy range from 60 eV to 200 eV. By comparing the experimental kz dispersion to the DFT band structure, we identified 129 eV and 115 eV photon energies measuring the kz ≈ 0 and kz ≈ π high-symmetry planes, respectively. The data in Fig. 2 are acquired with 129 eV photons, while the data in Fig. 3 are measured with 115 eV photons. All normal state (charge-ordered state) data in the main text is obtained at 120 K (6 K) using linear horizontal light polarization, unless specified. We refer to Supplementary Fig. 9 for the data measured in finer temperature steps.
Spatially resolved ARPES and XPS experiments
The real-space map**s of the valence band structure and core level spectra were conducted at Beamline 7.0.2 (MAESTRO) of the Advanced Light Source. To resolve the complex surface domains of ScV6Sn6, we used the micro-focused synchrotron of lateral dimension 30 × 30 μm2. The domain dependent ARPES and XPS spectra are compared to the slab DFT calculations of various geometries, to assign the atomic termination layer to each domain (Supplementary Note 2, 3).
DFT calculations
DFT calculations were performed using the Vienna AB initio Simulation Package software47,48. The generalized-gradient approximation Perdew-Burke-Ernzerhof exchange-correlation functional was chosen to calculate the exchange-correlation energy49. The pseudopotential was defined based on the projector augmented-wave method50. VASPKIT software was used for pre- and post-processing of DFT calculated information51. For the bulk band calculation of ScV6Sn6, we used the lattice parameters (a, b, c) = (5.456 Å, 5.456 Å, 9.230 Å) which is obtained by relaxing the reported single crystal refinement data37. Relaxation is performed at the 350 eV kinetic energy cutoff that fully covers the atomic energy. The static electronic structure was calculated using a Γ-centered k-point mesh, 15 × 15 × 8 for the normal state structure and 8 × 8 × 3 for the charge ordered structure. We present the overall band dispersion of ScV6Sn6 in the normal state with and without spin-orbit coupling in Supplementary Fig. 10a. Supplementary Fig. 10b displays the unfolded DFT band dispersion in the charge ordered state. To understand the termination-dependence of the valence band and core level spectra, we performed the slab DFT calculation on all possible charge neutral slab configurations of ScV6Sn6 relaxed at the 350 eV kinetic cutoff energy (Supplementary Note 2, 3). Γ-centered 11 × 11 × 1 k-point mesh were used for the slab band calculation. Each slab has 20 atomic layers and the vacuum was fixed at 20 Å.
DFPT calculations
Phonon dispersions were computed within the density functional perturbation theory (DFPT) framework. Input parameters were generated from the 3 × 3 × 2 supercell using Phonopy software52,53. We compared the phonon modes of two kagome metals, ScV6Sn6 and CsV3Sb5. Various smearing factors 0.10, 0.125, 0.15, 0.175, and 0.20 were tested for both compounds. We present the smearing factor-dependent phonon dispersions in Supplementary Fig. 11.