
Abstract
Understanding the durability of nuclear glass in the vapor phase is essential for evaluating the long-term safety of the nuclear glass. This work studies the vapor hydration of borosilicate and iodine-bearing glass at 90 °C and relative humidity at 99%. Hydration kinetics was monitored by analyzing the boron and iodine released during the vapor hydration process. It is found that boron becomes more easily released after iodine being added to the glass matrix. The results related to boron release and retention indicate the possible transformation of tetrahedral [4]B to trigonal [3]B on the hydrated glass surface, which may enhance the release of boron in the form of boric acid afterward. Release of iodine occurred but at a far lesser extent than boron with 92% of iodine retained in the alteration layer, thus opening good perspectives with regard to the disposal of high-level waste containing iodine.
Similar content being viewed by others


Introduction
Highly radioactive and long-lived waste arising from the reprocessing of spent nuclear fuel is vitrified in the form of borosilicate nuclear glass in France. It is planned to dispose of the nuclear glass permanently in the underground geological disposal facilities to ensure the isolation of radionuclides from human beings and the environment. Under the context of disposal cell re-saturation due to the groundwater penetration, the durability of nuclear waste glass in saturated media (or in aqueous solution) has been largely assessed during the past three decades by studying the dissolution of nuclear glasses as a function of pH, temperature, solution composition, and glass composition, as well as external and internal irradiation effects1,2,3,4,5. However, before the disposal cell re-saturation, studies show that the geological disposal cell may keep unsaturated for tens of thousands of years due to the presence of hydrogen gas generated via the corrosion of iron canister6,7. Thus, it is probable that nuclear glass will contact water vapor prior to the aqueous solution. As a result, special attention has been drawn to the hydration of nuclear glass under unsaturated vapor conditions in the last ten years8,9,10,11,12,13,14.
Early studies concerning the hydration of nuclear glass in the vapor phase started in the 1980s in the USA15,16,17,18,19. In their studies, most nuclear glass vapor hydration tests were performed at a high temperature varying from 120 to 260 °C and saturated steam condition (relative humidity–RH at 100%); the hydration rate has been estimated by the increase of the hydrated layer thickness observed by Scanning Electron Microscopy (SEM). From the 2010s, studies on nuclear glass hydration performed in France focus on the unsaturated vapor conditions (RH < 100%) at a temperature ranging from 35 to 200 °C8,9,10,11,12,13,20. The experiments were conducted in stainless steel autoclaves where the RH was controlled by the addition of saline solution in the Telon liner (Fig. 1). The vapor hydration kinetics have been followed by a non-destructive method based on the analyses of the glass samples by Fourier Transformed Infrared Spectroscopy (FTIR) performed at each sampling during the vapor hydration process8. Different parameters affecting the hydration kinetics have been investigated, including temperature, RH, pH, and glass composition8,9,10,11,12,13,20. A recent review on glass hydration in the vapor phase can be found in the Springer Handbook of Glass21. However, compared to the numerous studies on glass dissolution in the aqueous phase, the understanding of the glass vapor hydration mechanisms is still limited.

Experimental setup developed at SUBATECH Laboratory.
In the current work on nuclear glass vapor hydration, special attention was given to two key elements: boron and iodine. Boron is usually considered a good tracer for the dissolution of nuclear glass in aqueous conditions22,23,24. Moreover, boron, as a crosslinking element within the aluminosilicate network, is considered to play an important role in the formation and reorganization of the alteration layer during glass aqueous dissolution25,26. Iodine is known for its volatility at relatively low temperatures, including room temperature27,28. In the studies of nuclear glass, the radionuclide 129I draws attention due to its high mobility in the environment and its major contribution to radioactive dose release from disposal site29. Furthermore, in the past work on glass vapor hydration, it has been shown that the surface of the vapor-hydrated glass is significantly depleted in boron without any explanation being put forward13. We believe that iodine, known for its volatility, could suffer the same fate with the possibility of ending up in the saline solution. Hence, to study the behavior of boron and iodine during the glass vapor hydration process, a method (denoted ‘chemical method’ in this study) based on the quantification of boron and iodine deposited in the saline solution in the autoclave—Fig. 1) was proposed and verified by performing vapor hydration tests with chemical compounds containing boron or iodine and with duplicate borosilicate glass samples. The chemical method was then applied to follow the hydration kinetics of the iodine-bearing borosilicate glasses. A well-established method8 (denoted ‘physical method’ in this study) based on the solid characterization by FTIR was also applied as a reference to study the glass hydration kinetics. By comparing the boron and iodine releases between glass samples with similar compositions and by performing the glass characterizations, discussions on the effect of iodine addition in glass structure change are given, which gives insights into the mechanism of boron release in return.
Results
Feasibility of chemical method to follow the hydration kinetics
To study the phenomenon of glass depletion in boron and iodine, we tested the behavior of chemical compounds containing boron and iodine under the vapor hydration condition. Vapor hydration tests with five chemical compounds (H3BO3, NaBO2.4H2O, Na2B4O7, NaI, and NaIO3) were performed at 90 °C in the autoclave that contains 8 mL of saline solution (1.75 wt% of NaCl ensuring 99% of RH). We intended to test if boron or iodine could be volatilized under the experimental vapor condition and if the volatilized elements could be quantified by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). After 3 days of vapor hydration, the ICP-MS analysis results showed that boron and iodine could be successfully quantified in the saline solution, which confirmed their volatility from the tested chemical compounds under the experimental vapor condition. After 28 days of vapor hydration, 16.3 ± 1.2% of H3BO3 (compared to its initial quantity) were found to be deposited in the saline solution, with 1.6 ± 0.1% of Na2B4O7 and less than 1% of NaBO2·4H2O, NaI, and NaIO3.
The tests with the chemical compounds demonstrated the feasibility of quantifying volatilized boron and iodine under the vapor condition. The idea of boron quantification was further developed into the chemical method through which the glass hydration kinetics could be deduced from the boron release. The application of the chemical method requires regular sampling of the saline solution. The influence of sampling, typically the heating and cooling cycle, was tested by performing samplings of different time intervals with two identical glass samples.
Duplicate NH glass samples (Composition given in Table 1)30, NH-1 and NH-2, were hydrated at 90 °C and RH at 99% for 28 days in the autoclaves. Four samplings were performed with NH-1 after 7, 14, 21, and 28 days. Only one sampling was performed with NH-2 at the end of 28 days of vapor hydration. At each sampling, the saline solution was renewed and the used solution was collected for ICP-MS analysis. Si and Al concentrations were too low to be detected by ICP-MS. Thus, only B was quantified for the tests with NH samples. The limit of quantification (LoQ) is 80 µg L−1 for Al, 1320 µg L−1 for Si, and 11 µg L−1 for B. The saline solutions were diluted 200 times before analysis to decrease the solutions’ salinity.
Figure 2 gives the variations of the normalized mass loss calculated from boron release (NLB) for NH-1 and NH-2. The NLB for NH-1 was calculated based on the cumulated boron quantity obtained after each sampling. After 28 days of vapor hydration, the NLB of NH-1 was slightly lower than that of NH-2. The difference of NL could be due to the perturbation of the heating and cooling cycles during the sampling, but it remains within the uncertainties. The vapor hydration tests with two NH samples confirmed that the samplings (heating and cooling) during the glass vapor hydration process would not stop the boron release; the perturbation of sampling could be neglected.

Variation of NLB of NH-1 and NH-2 glass samples hydrated at 90 °C and RH at 99% with different sampling intervals: 4 samplings for NH-1 glass, 1 sampling for NH-2 glass. The error bars represent the standard deviation.
Glass hydration followed by physical and chemical methods
Three glass samples, ISG, ISG33, and VNaI (Compositions are given in Table 1) have been hydrated in autoclaves at 90 °C with 99% RH. The physical method based on FTIR measurements and the chemical method based on saline solution analysis (for B and I concentrations) were applied to ISG and VNaI samples to study the vapor hydration kinetics. In the chemical method, the chemical thickness was calculated based on the analysis of boron concentration in the saline solution collected from each sampling. In the physical method, a converting coefficient of 0.11 ± 0.02 Abs μm−1, which converts the increase of Si–OH absorbance (obtained by infrared (IR) spectra deconvolution) to the increase of the hydrated layer thickness, has been determined for ISG samples in the previous study9. For VNaI sample, its converting coefficient was determined by coupling the cross-section observation by SEM (Fig. 3) and IR spectrum analysis. At the end of 108 days of vapor hydration, the hydrated layer thickness of VNaI sample was found to be 1.59 ± 0.24 μm by observing its cross-section via SEM (Fig. 3). The deconvoluted IR spectrum obtained from the last sampling showed an increase of 0.19 ± 0.02 in Si–OH absorbance compared with that of the pristine glass spectrum. It could be calculated that the converting coefficient of VNaI samples is 0.12 ± 0.02 Abs μm−1. The variations of the Si–OH absorbance obtained during the vapor hydration process could then be converted to the variations of the hydrated layer thickness (physical thickness) by using the converting coefficients of ISG and VNaI.

SEM cross-section of VNaI sample hydrated for 108 days at 90 °C and RH at 99%.
Figure 4 gives the variations of physical and chemical thicknesses of ISG and VNaI samples. A similar tendency of increase between physical and chemical thicknesses could be observed for both ISG and VNaI. However, for both samples, the chemical thickness is always smaller than the corresponding physical thickness. The variations of the ratio between chemical and physical thickness (Rc/p) are given in Fig. 4a, b. After about 40 days of vapor hydration, Rc/p stabilized at about 0.66 for ISG and 0.47 for VNaI. After 150 days of vapor hydration of ISG, the difference between physical and chemical thickness reduced, and Rc/p increased to 0.8.

Comparison of hydrated layer thickness estimated by physical and chemical methods: a, b variation of Rc/p of hydrated ISG and VNaI; c, d variation of the chemical and physical thickness of hydrated ISG and VNaI. Chemical thickness was deduced based on the quantity of boron found in the saline solutions. The error bars represent the standard deviation.
The difference between chemical and physical thicknesses could be explained by the retention of boron in the hydrated layer. The equivalent chemical thickness calculation assumed that 100% of boron within the hydrated layer was released. Thus, the chemical thickness of the hydrated layer could be underestimated if part of the boron was still retained in the hydrated layer.
Comparison of boron and iodine release
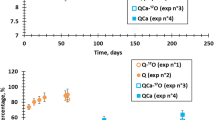
The releases of boron and iodine (represented by the normalized mass loss, NL) from VNaI and ISG33 are given in Fig. 5. Figure 5a shows the variations of ratios between NLI and NLB of ISG33 and VNaI. No congruent release between boron and iodine could be observed in Fig. 5b. It could be seen that iodine is significantly retained in VNaI compared with ISG33. By converting the NLI to the equivalent thickness then comparing it with the physical thickness, it could be calculated that about 9% of iodine was deposited in the saline solution while the rest of 91% is likely to be retained in the hydrated layer.

Comparison of boron and iodine release from ISG33 and VNaI glass samples hydrated at 90 °C and RH at 99%: a variation of NLB/NLI ratio of hydrated ISG33 and VNaI; b variation of NLB and NLI of hydrated ISG33 and hydrated VNaI. The error bars represent the standard deviation.
Figure 6 gives the release of boron from ISG and ISG33 glass samples, which have similar compositions with only ISG33 doped with iodine. From ISG33, more boron was released than from the reference ISG. The effect of iodine addition on the release of boron is discussed in the “Discussion” section.

ISG and ISG33 were hydrated at 90 °C and RH at 99%. The error bars represent the standard deviation.
Hydrated surface characterization
A duplicate of the VNaI sample was hydrated for 126 days at 90 °C with 99% RH. The hydrated sample was analyzed by Time-of-Flight Secondary Ion Mass Spectroscopy (ToF-SIMS) to obtain elemental profiles and hydrated layer thickness. Figure 7 gives the normalized profiles of H, B, Na, and I. Considering the large quantify of Si in the glass samples and that Si was not found in saline solution, the concentration of Si could be assumed as unchanged in the hydrated layer. Thus, all the data in Fig. 7 were firstly normalized to the relative intensity by dividing the Si intensity in the hydrated layer (C/CSi), then to the relative mean intensity in the pristine glass, denoted as PG \(\left( {{\mathrm{normalized}}\,{\mathrm{intensity}} = \frac{{{\mathrm{C/}}C_{{\mathrm{Si}}}}}{{\left( {\frac{C}{{C_{{\mathrm{Si}}}}}} \right)_{{\mathrm{PG}}}}}} \right)\). The hydrated layer thickness is determined to be 0.83 µm based on the boron profile (normalized intensity of boron = 0.5). The error of the estimated thickness value could be considered as lower than 10% based on similar SIMS studies on the other glasses31,32. The estimated physical thickness is 0.88 ± 0.15 µm based on the FTIR analysis. The FTIR estimation is slightly larger than the SIMS estimation, but the difference is still within the uncertainties. Besides, the physical thickness was calculated by considering the intensity of Si-OH. The different elements used for FTIR and SIMS estimations (Si–OH and B, respectively) may cause the difference in the thickness estimation.

VNaI glass sample was hydrated for 126 days at 90 °C and RH at 99%.
The depletion of boron and sodium in the hydrated layer could be observed. By calculating the surface area under the curve of the normalized boron profile, we find that about 9% of boron (initially present in the glass) was retained in the hydrated layer. The chemical thickness is calculated to be 0.74 ± 0.07 µm (based on the quantity of boron release). The ratio between the chemical thickness and the SIMS estimated thickness (0.83 µm) is 0.89, which indicates a boron retention ratio of 11%. The coherence between the two estimations of boron retention ratio demonstrates that almost all the released boron has been recovered in the saline solution then analyzed by ICP-MS. The chemical method by ICP-MS measurements coupled with SIMS analysis could be a reliable method for vapor hydration kinetics study. Besides, about 30% of Na is retained in the hydrated layer. A nearly constant level of Na is also observed in the hydrated layer.
The depletion of iodine could also be observed, but only in the first 360 nm. About 92% of iodine is retained in the hydrated layer. The strong retention of I is also coherent with the observations concerning the B and I release quantified by ICP-MS.
Micro-Raman analysis has been performed to characterize the glass surface of ISG and VNaI before and after vapor hydration. Figure 8 shows the spectra in the region between 200 and 1700 cm−1. Spectra of hydrated ISG and VNaI were normalized to the maximum intensity at around 500 cm−1 of the corresponding pristine glass spectra. The broad peak at around 500 cm−1 is attributed to mixed stretching and bending vibrational modes of Si–O–Si and Si–O–B33,34. The narrow peak at 630 cm−1 could be assigned to the breathing mode of danburite rings (B2O7–Si2O7)35. Several vibrational bands could be attributed in the region between 650 and 820 cm−1, including tetraborate groups at 670 cm−1, four-coordinated boron in diborate rings at 770 cm−1, and boroxol rings at 808 cm−134,36,37. Compared with the spectra of the pristine glasses, a slight decrease in the contribution of four-coordinated boron at 670 and 770 cm−1 could be observed through the decrease of the intensity in this region. However, it is difficult to quantify the decrease of [4]B from the Raman spectra. The region between 1300 and 1600 cm−1 is considered as a typical region of three-coordinated boron37. A narrow peak located at 1350 cm−1 could be observed on the hydrated samples. A similar peak at 1350 cm−1 has been assigned to the asymmetric stretching of the three-coordinated B–O bond in the Raman study of hydrated borates33. The decrease in [4]B and the appearance of [3]B-O bond at 1350 cm−1 suggest the transformation from [4]B to [3]B in the hydrated layer.

Comparison of Raman spectra between pristine ISG, hydrated ISG (412 days, 90 °C, RH at 99%), pristine VNaI, and hydrated VNaI (126 days, 90 °C, RH at 99%) glass samples. The baseline of Raman spectra is subtracted with the third-order polynomial function. Refer to the “Methods” section for more information on baseline correction.
Based on various Raman studies of alkali silicate, borosilicate, or aluminoborosilicate glasses, the broad peak between 850 and 1200 cm−1 is usually considered to be composed of several Qn bands (n varies from 0 to 4) which denote Si–O– stretching in a structural unit with n non-bridging O atoms and 4-n bridging O atoms per silicon34,38,39,40. The Raman spectra of the pristine and hydrated glasses were fitted with four Gaussians in Origin 8.0© software by considering the following band assignments: Q1 band located between 880 to 930 cm−1, Q2 at around 980 cm−1, Q3 at around 1070 cm−1, and Q4 at around 1150 cm−134,38,39,40. Though there are still discussions on the band attribution in this region, it is a common method to study the glass structure change by looking at the variation of fitted peak area in the high-frequency region (typically Q3 and Q4) and in the low-frequency region (Q1 and Q2)34,38,39,41,42. The deconvolution of the hydrated and pristine glasses’ Raman spectra showed that the ratio between Q1 + Q2 and Q3 + Q4 (denoted as R12/34) has increased from 1 to 1.4 for the hydrated ISG, and from 0.26 to 0.39 for the hydrated VNaI (Fig. 9). The increase of R12/34 could be used to indicate the depolymerization of the glass network34. Thus, it could be concluded that the structure of the hydrated glass surface has been depolymerized, at least within the analytical depth (~2 µm). As Si and Al were strongly retained in the hydrated layer, the depolymerization of the hydrated layer could probably be attributed to the release of boron, which is also coherent with the study of Collin et al.25 where they suggested that boron played a role in crosslinking element in the aluminosilicate network.

a Deconvoluted Raman spectra of pristine and hydrated ISG (412 days, 90 °C, RH at 99%) samples; b deconvoluted Raman spectra of pristine and hydrated VNaI (126 days, 90 °C, RH at 99%) samples.
Discussion
The comparison of boron and iodine releases from different glass samples showed that more boron could be released when iodine was added to the glass matrix and that boron and iodine releases could be dependent on the glass composition. Studies on iodine speciation in borosilicate glasses show that iodide (I−) is the predominant form of iodine and surrounded preferentially by Na+ ions in the glass matrix43,44,45,46. The environment of sodium surrounding iodide was proposed to be precursors to the formation of iodosodalite (Na8(AlSiO4)6I2)45. The crystallization of iodosodalite has been observed when large quantities of iodine were added into glass matrix44: Iodosodalite has been considered as a potential waste form of iodine disposal47. The leaching tests of iodosodalite showed high chemical durability in natural underground water saturated with Si and Al48. Considering the possibility of possessing precursors to iodosodalite in a Na-rich glass environment and the chemical durability of iodosodalite, the high concentration of Na in VNaI glass could contribute to the fixation of iodine in the glass matrix, thus justifying the low release of iodine in the vapor phase.
The quantity of sodium in glass could also influence the speciation of boron in borosilicate glasses, which may further influence the behavior of boron release. In a glass network, sodium could play two roles: modifier of glass network or charge compensators to network formers49,50,51. When it acts as a network modifier, adding sodium into the glass will result in the formation of Non-Bridging Oxygens (NBOs) and the depolymerization of the glass network. When it acts as a charge compensator, Na+ is used to compensate for the negative charge of network formers units such as \({\mathrm{AlO}}_4^ -\) and \({\mathrm{BO}}_4^ -\). The changes in sodium role between network modifier and charge compensator have been discussed in several studies49,50,51. Considering the surrounding environment of iodine in the glass structure, when iodine is added to ISG33, sodium would be preferentially located at the position surrounding iodide ions, which may decrease the availability of sodium acting as a charge compensator for \({\mathrm{BO}}_4^ -\). As a result, the fraction of the four-coordinated boron should decrease in ISG33. In the study of Jolivet et al.52, the fraction of four-fold coordinated boron among boron species (N4, defined as [BO4]/([BO4] + [BO3])) was determined to be 0.5 for pristine ISG, and around 0.48 for iodine-bearing glass samples. Besides, for the iodine-bearing ISG samples, Jolivet et al. also showed that the high-pressure preparation process had a minor impact on the N4 compared with the impact of iodine addition52. Furthermore, considering that more boron has been released from ISG33 with a higher [3]B fraction, it may suggest that boron in the form of trigonal BO3 could be preferentially released. Micro-Raman characterization of the hydrated surface also showed the increase in the contribution of BO3 which could be ready to release from the surface of the hydrated layer in our experimental conditions. It is therefore possible that boron is evaporated in the form of H3BO3 from the glass surface. Besides, it should be noted that ISG sample was polished to 3 µm before the vapor hydration test while ISG33 was not polished due to its small size. As revealed in the studies concerning the effect of initial surface state53,54, the important surface roughness could also contribute to the large release of boron from ISG33 than from ISG sample.
When testing the feasibility of the chemical method, the vapor hydration tests with 3 types of chemical powders containing boron (H3BO3, Na2B4O7, and NaBO2·4H2O) showed that H3BO3 presented higher volatility than Na2B4O7 and NaBO2·4H2O. The evaporation of boric acid from seawater at ordinary temperature has been confirmed by Gast and Thompson55. The strong volatility of boron from acid solution at a higher temperature (65–70 °C) was reported by Ishikawa and Nakamura56. All these studies showed strong volatility of H3BO3.
In a recent study of borate glass alteration in liquid water, Goetschius et al.57 also observed that the minimum glass dissolution rates occurred with the sample possessing the maximum tetraborate fraction. The study on the hydrolysis of borosilicate glass showed that in an aqueous environment, the bridging bond of tetrahedral [4]B with Si or B would break to form a trigonal [3]B and a Si-OH or a B-OH group58. Goetschius et al.57 also proposed that boron was released in the form of boric acid into the leaching solution. Although our experimental conditions are different, the outer surfaces of our samples were covered with several layers of water molecules, allowing hydrolysis of B-O bonds to occur. Thus, a similar hydrolysis process could happen to transform the [4]B to [3]B.
Unlike the nearly total depletion of Na in the alteration layer of glass obtained in the studies of glass aqueous alteration59,60,61, the Na profile in the hydrated VNaI showed that 30% of Na (3.25 mol%) was retained in the hydrated layer. The retained Na could probably still play the role of charge compensator for I−, \({\mathrm{AlO}}_4^ -\), and \({\mathrm{BO}}_4^ -\). Based on the elemental profiles in the hydrated layer of VNaI (Fig. 7), we showed that 92% of I and 9% of B were retained in the hydrated layer, which corresponds to the concentrations of 0.24 mol% and 0.86 mol% for I and B, respectively. By supposing that the quantity of Al (2.15 mol%) stayed unchanged during the vapor hydration of VNaI and that all the Al in the hydrated layer is in the form of \({\mathrm{AlO}}_4^ -\) (the predominance of tetrahedral Al was found in gel layer resulting from the aqueous alteration62), it requires 2.39 mol% of Na+ to obtain the charge balance with I− and \({\mathrm{AlO}}_4^ -\). Then 0.86 mol% of Na in excess could be used to compensate the charge of \({\mathrm{BO}}_4^ -\) in the hydrated layer, which indicates that nearly all the retained B are in the form of \({\mathrm{BO}}_4^ - .\) However, it should be noted that the results of the previous calculation could be influenced by the surrounding environment of I− in the hydrated layer (I could be surrounded by several Na)43,44,45,46. The constant level of Na distribution in the hydrated layer may indicate a stable and homogeneous structure of the hydrated layer. As discussed in the previous paragraphs, we proposed that B would firstly transform from [4]B to [3]B before its release. Near the interface of the hydrated layer and pristine glass (between 360 to 820 nm in Fig. 7), the levels of Na and I are constant, which may lead to a constant level of [4]B. Thus, the B in excess in this region could be in the form of [3]B and ready to diffuse towards the glass surface (glass/vapor interface). In the recent study of the aqueous alteration of ISG carried out by Gin et al, they also showed the diffusion of soluble B(aq) species towards the glass, which may be a rate-limiting factor for the growth of alteration gel due to the glass network reorganization26.
Hydration of borosilicate glasses doped with iodine has been studied under vapor conditions. The glass hydration kinetics is studied by monitoring the quantities of released boron and iodine during the alteration in the vapor phase. The comparison of boron release between different glass samples showed that boron in the form of three-coordinated [3]B could be preferentially released. The distribution of [3]B and [4]B is influenced by the availability of sodium in the boron surrounding environment, which is strongly dependent on the iodine content in the glass. The characterization of the hydrated glass surface also showed evidence of boron form transformation from [4]B to [3]B. The behavior of B needs to be further studied to determine if it could play a similar role in limiting the hydration kinetics as indicated in the study of glass alteration in the aqueous phase26.
By comparing the physical thickness (obtained from FTIR), the thickness given by ToF-SIMS, and the chemical thickness (derived from the deposited boron concentration in the saline solution), about 9 to 40% of boron and up to 92% of iodine were found to be retained in the hydrated layer. The strong retention of iodine in the hydrated layer is very important in terms of radiological impact under the context of nuclear waste storage. However, the speciation of iodine in the hydrated layer still needs to be investigated to better assess its stability and retention properties. Furthermore, it is also necessary to study the alteration of hydrated glass in aqueous conditions, in the prediction of the re-saturation of the disposal cells with groundwater.
Methods
Sample preparation
To test the behaviors of boron and iodine during the vapor hydration process, hydration tests were performed with five types of commercial chemical powder containing either boron or iodine: H3BO3 (Sigma-Aldrich, ≥99.5%), NaBO2.4H2O (Sigma-Aldrich, ≥99%), Na2B4O7 (Sigma-Aldrich, 99%), NaI (Sigma-Aldrich, 99.999%), and NaIO3 (Fisher Scientific, ≥99%).
Glass samples of four compositions (Table 1) were used for the glass vapor hydration tests. The International Simple Glass (ISG) was produced in 2012 by MoSci Corporation (Rolla, MO, USA). ISG is considered as a reference borosilicate nuclear glass that is composed of six oxides63. The ratios between the six oxides are the same as those between the French inactive reference nuclear glass SON681. Glass monoliths of ISG with a dimension of 20 × 20 × 1 mm3 were cut. VNaI glass was also provided by CEA. Glass monoliths of VNaI with a dimension of 10 × 5 × 1 mm3 were cut. The ISG and VNaI glass monoliths were polished with SiC paper to grade 4000 and with diamond suspension (3 μm), then cleaned with acetone in an ultrasonic bath before use. The glass sample of ISG33 was provided by the Laboratoire de Planétologie et Géodynamique de Nantes (LPG-Nantes). It is an I-bearing glass synthesized under high temperature 1500 °C and high-pressure conditions at 1.5 GPa52. The quantity of ISG33 was very limited and the polishing of ISG33 was not able to be performed. ISG33 was cleaned with acetone in an ultrasonic bath before use. The surface area of ISG33 was estimated geometrically with the help of a Leica MZ16 stereomicroscope. Another glass sample, NH, provided by LPG-Nantes52 was used to verify the experimental method (chemical method) used in this study.
Experimental set-up
All the vapor hydration tests have been conducted in the stainless-steel autoclave with a Teflon liner (Fig. 1). The experimental set-up is similar to the ones used in the previous studies performed in Subatech laboratory8,9. A Teflon liner with an inner volume of 39 mL was put inside the autoclave. To control the relative humidity in the Teflon liner, 8 mL of saline solution (NaCl, Sigma-Aldrich, 99.999%) was placed in the bottom. The concentration of the saline solution is 1.75 wt% to maintain the relative humidity at 99%64. The sample was placed on a Teflon holder, to keep the sample separated from the 8 mL of saline solution. The assembled and tight autoclave was then placed in a 2 cm thick aluminum container. The whole set-up was then kept in an oven at 90 °C for temperature control during the vapor hydration test. At each sampling, the autoclave was cooled down to room temperature in the aluminum container before being opened.
For vapor hydration tests with glass samples, periodic samplings were performed to follow the kinetics of boron and iodine releases throughout the vapor hydration process. The sampling time intervals ranged between 7 days to 24 days with shorter intervals at the beginning of the runs. At each sampling, the 8 mL of saline solution was collected for boron and iodine concentration analysis and renewed by a fresh solution. We collected for each experiment at least 6 aliquots for analysis.
Experimental techniques
Boron and iodine concentrations have been analyzed by inductively coupled plasma mass spectrometry (Quadrupole ICP-MS Xseries 2, Thermoelectron). Boron was analyzed in HNO3 with beryllium as an internal standard. Iodine was analyzed in NH4OH with indium as an internal standard. The limits of quantification of ICP-MS are 0.054 μg·L−1 for boron and 0.012 μg·L−1 for iodine with relative uncertainties at 5–10%.
At each sampling, the glass monoliths were analyzed by FTIR Spectroscopy (Bruker-Tensor 27) at room temperature. IR spectra were collected in the range of 2500 to 4000 cm−1 with 1 cm−1 resolution. 32 scans were taken and averaged for each spectrum. The spectrum of the background was taken then subtracted automatically in OPUS© software. The FTIR analysis allowed the deduction of the alteration layer thickness based on a method developed by Neeway et al.8.
The cross-section of the VNaI hydrated samples was prepared to allow layer thickness measurement and characterization with a Scanning Electron Microscope JEOL JSM 5800LV. The operating voltage was 15 kV. The VNaI glass sample was glued on the SEM specimen holders with a conductive coating material (PELCO® Conductive Silver 187).
The elemental profiles in the hydrated layer of VNaI were obtained by Time-of-Flight Secondary Ion Mass Spectroscopy (ToF-SIMS). The profiles of negative secondary ions were obtained by alternating analysis and abrasion cycles. Primary ions of \({\mathrm{Bi}}_3^ +\) were applied for abrasion cycles at 25 keV and with the current at 0.3 pA. The analyzed area was 50 × 50 µm2.
The glass surface was analyzed by micro-Raman spectroscopy to identify the formed hydration products. The Raman spectra were acquired using a Jobin-Yvon Labram© HR800 Evolution spectrometer equipped with 300 lines per mm diffraction gratings corresponding to a spectral resolution of ~3 cm−1. A 100× Olympus objective was used for sample analysis. The spectra were acquired with a solid-state 532 nm wavelength laser with output power at 50 mW preventing the destruction of the secondary hydration products. A confocal mode was used to obtain a 2 µm depth excitation spot at the samples. Raman spectra were acquired between 200 and 1800 cm−1. Typical spectra were acquired for 60 s and 2 repetitions were achieved to obtain a good signal-to-noise ratio. Raman spectra were background-subtracted with 3rd order polynomial function by considering four anchor points in the no-band areas: ~200, ~850, ~1250, and ~1700cm−1.
Chemical methods used to follow hydration kinetics
Hydration kinetics could be followed by investigating the variation of hydrated layer thickness. In the study of glass alteration in the aqueous phases8, an equivalent thickness (Thc) of the altered layer could be calculated by applying the following equations:
NLi is the normalized mass loss (NL, g·m−2) calculated from the release of element i. Thc is the equivalent thickness, with ρ the density of the glass sample. Ci represents the concentration of element i. fi is the mass fraction of element i in the pristine glass. S is the surface area of the glass sample. V is the volume of saline solution (8 mL) used for relative humidity control. For a practical reason, we call the thickness estimated from the release of boron ‘chemical thickness’.
The physical method used to follow hydration kinetics
Glass hydration kinetics were followed by FTIR analysis coupled with SEM observation, which is denoted as the ‘physical method’ in this study. The physical method has been largely used and described in recent studies8,9,10,11,12,13,20. The IR spectra obtained from 2500 to 4000 cm−1 could be fitted with five Gaussian bands:
-
~2700 cm−1 attributed to silica matrix;
-
~3170–3185 cm−1 attributed to OH stretching mode in bound water silanol groups;
-
~3400–3415 cm−1 attributed to OH symmetrical stretching mode in free water molecule;
-
~3515–3518 cm−1 attributed to OH stretching mode in bound water silanol groups;
-
~3595–3605 cm−1 attributed to OH stretching mode in Si-OH.
An example of IR spectrum deconvolution is given in Fig. 10. The deconvolution method and vibrational bands assignment could be referred to as the study of water-related bands in glasses performed by Efimov et al. and Navarra et al.65,66. The increase of Si-OH band absorbance (~3600 cm−1) has been shown to be proportional to the increase in the hydrated layer thickness8,9,20. With the help of SEM observation of the hydrated layer, the coefficient that converts the increase in Si-OH absorbance and the hydrated layer thickness (physical thickness) could be determined. The variation of the hydrated layer thickness during the vapor hydration process could then be deduced.

The increase of Si–OH band absorbance is considered to be proportional to the increase in the hydrated layer thickness. Refer to the “Methods” section for more information on IR spectra deconvolution.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Gin, S. et al. An international initiative on long-term behavior of high-level nuclear waste glass. Mater. Today 16, 243–248 (2013).
Peuget, S., Delaye, J. M. & Jégou, C. Specific outcomes of the research on the radiation stability of the French nuclear glass towards alpha decay accumulation. J. Nucl. Mater. 444, 76–91 (2014).
Peuget, S., Tribet, M., Mougnaud, S., Miro, S. & Jégou, C. Radiations effects in ISG glass: from structural changes to long-term aqueous behavior. npj Mater. Degrad. 2, 23 (2018).
Vienna, J. D., Ryan, J. V., Gin, S. & Inagaki, Y. Current understanding and remaining challenges in modeling long-term degradation of borosilicate nuclear waste glasses. Int. J. Appl. Glas. Sci. 4, 283–294 (2013).
Frankel, G. S. et al. A comparative review of the aqueous corrosion of glasses, crystalline ceramics, and metals. npj Mater. Degrad. 2, 15 (2018).
El Mendili, Y., Abdelouas, A., Ait Chaou, A., Bardeau, J. F. & Schlegel, M. L. Carbon steel corrosion in clay-rich environment. Corros. Sci. 88, 56–65 (2014).
ANDRA. Tome Évolution phénoménologique du stockage géologique. Dossier 2005 Argile (2005).
Neeway, J. et al. Vapor hydration of SON68 glass from 90 °C to 200 °C: A kinetic study and corrosion products investigation. J. Non Cryst. Solids 358, 2894–2905 (2012).
Abdelouas, A. et al. A preliminary investigation of the ISG glass vapor hydration. Int. J. Appl. Glas. Sci. 4, 307–316 (2013).
Ait Chaou, A. et al. Vapor hydration of a simulated borosilicate nuclear waste glass in unsaturated conditions at 50 °C and 90 °C. RSC Adv. 5, 64538–64549 (2015).
Ait Chaou, A., Abdelouas, A., El Mendili, Y. & Martin, C. The role of pH in the vapor hydration at 175 °C of the French SON68. Glass Appl. Geochem. 76, 22–35 (2017).
Bouakkaz, R., Abdelouas, A. & Grambow, B. Kinetic study and structural evolution of SON68 nuclear waste glass altered from 35 to 125 °C under unsaturated H2O and D2O18 vapour conditions. Corros. Sci. 134, 1–16 (2018).
Narayanasamy, S. et al. Influence of composition of nuclear waste glasses on vapor phase hydration. J. Nucl. Mater. 525, 53–71 (2019).
Majérus, O. et al. Glass alteration in atmospheric conditions: crossing perspectives from cultural heritage, glass industry, and nuclear waste management. npj Mater. Degrad. 4, 27 (2020).
Bates, J. K., Jardine, L. J. & Steinler, M. J. The Hydration Process of Nuclear-waste Glass: an Interim Report. ANL–82-11 (Argonne National Laboratory, Argonne, IL, 1982).
Bates, J. K., Steindler, M. J., Tani, B. & Purcell, F. J. The hydration alteration of a commercial nuclear waste glass. Chem. Geol. 51, 79–87 (1985).
Abrajano, T. A. Jr, Bates, J. K. & Byers, C. D. Aqueous corrosion of natural and nuclear waste glasses I. Comparative rates of hydration in liquid and vapor environments at elevated temperatures. J. Non-Cryst. Solids 84, 251–257 (1986).
Abrajano, T. A., Bates, J. K. & Mazer, J. J. Aqueous corrosion of natural and nuclear waste glasses II. Mechanisms of vapor hydration of nuclear waste glasses. J. Non Cryst. Solids 108, 269–288 (1989).
Bates, J. K., Ebert, W. L. & Gerding, T. J. Vapor Hydration and Subsequent Leaching of Transuranic-Containing SRL and WV Glasses. In International High-Level Radioactive Waste Management Conference (United States, 1990).
Ait Chaou, A. et al. The French SON68 glass vapor hydration under different atmospheres. Procedia Mater. Sci. 7, 179–185 (2014).
Abdelouas, A., Neeway, J. & Grambow, B. Chemical durability of glasses. In Springer Handbook of Glass (eds. Musgraves, J. D., Hu, J. & Calvez, L.) (Springer International Publishing, 2019).
Frugier, P. et al. SON68 nuclear glass dissolution kinetics: current state of knowledge and basis of the new GRAAL model. J. Nucl. Mater. 380, 8–21 (2008).
Gin, S., Beaudoux, X., Angéli, F., Jégou, C. & Godon, N. Effect of composition on the short-term and long-term dissolution rates of ten borosilicate glasses of increasing complexity from 3 to 30 oxides. J. Non Cryst. Solids 358, 2559–2570 (2012).
Rajmohan, N., Frugier, P. & Gin, S. Composition effects on synthetic glass alteration mechanisms: Part 1. Experiments. Chem. Geol. 279, 106–119 (2010).
Collin, M. et al. Structure of International Simple Glass and properties of passivating layer formed in circumneutral pH conditions. npj Mater. Degrad. 2, 4 (2018).
Gin, S. et al. Insights into the mechanisms controlling the residual corrosion rate of borosilicate glasses. npj Mater. Degrad. 4, 41 (2020).
Lin, C. C. Volatility of iodine in dilute aqueous solutions. J. Inorg. Nucl. Chem. 43, 3229–3238 (1981).
Fuge, R. The role of volatility in the distribution of iodine in the secondary environment. Appl. Geochem. 5, 357–360 (1990).
ANDRA. Tome Évaluation De Sûreté Du Stockage Géologique. Dossier 2005 Argile (2005).
Jolivet, V. et al. Quantification of boron in aluminoborosilicate glasses using Raman and 11 B NMR. J. Non Cryst. Solids 511, 50–61 (2019).
Py, M. Study of Interfaces and Nanometric Structures by ToF-SIMS: upon A Spatially Resolved Quantitative Analysis. (Université de Grenoble, 2011).
Gin, S. et al. Dynamics of self-reorganization explains passivation of silicate glasses. Nat. Commun. 9, 1–9 (2018).
Li, J., **a, S. & Gao, S. FT-IR and Raman spectroscopic study of hydrated borates. Spectrochim. Acta Part A Mol. Spectrosc. 51, 519–532 (1995).
Manara, D., Grandjean, A. & Neuville, D. R. Advances in understanding the structure of borosilicate glasses: a Raman spectroscopy study. Am. Mineral. 94, 777–784 (2009).
Bunker, B. C., Tallant, D. R., Kirkpatrick, R. J. & Turner, G. L. Multinuclear nuclear magnetic resonance and Raman investigation of sodium borosilicate glass structures. Phys. Chem. Glas. 31, 30–41 (1990).
Yadav, A. K. & Singh, P. A review of the structures of oxide glasses by Raman spectroscopy. RSC Adv. 5, 67583–67609 (2015).
Meera, B. N., Sood, A. K., Chandrabhas, N. & Ramakrishna, J. Raman study of lead borate glasses. J. Non Cryst. Solids 126, 224–230 (1990).
Bonfils, Jde et al. Effect of chemical composition on borosilicate glass behavior under irradiation. J. Non Cryst. Solids 356, 388–393 (2010).
Neuville, D. R. Viscosity, structure and mixing in (Ca, Na) silicate melts. Chem. Geol. 229, 28–41 (2006).
Mysen, B. O. & Frantz, J. D. Silicate melts at magmatic temperatures: in-situ structure determination to 1651 C and effect of temperature and bulk composition on the mixing behavior of structural. Contrib. Mineral. Petrol. 117, 1–14 (1994).
Bauer, U., Behrens, H., Reinsch, S., Morin, E. I. & Stebbins, J. F. Structural investigation of hydrous sodium borosilicate glasses. J. Non Cryst. Solids 465, 39–48 (2017).
McKeown, D. A., Muller, I. S., Buechele, A. C., Pegg, I. L. & Kendziora, C. A. Structural characterization of high-zirconia borosilicate glasses using Raman spectroscopy. J. Non Cryst. Solids 262, 126–134 (2000).
Cicconi, M. R. et al. Iodine solubility and speciation in glasses. Sci. Rep. 9, 7758 (2019).
Riley, B. J. et al. Iodine solubility in a low-activity waste borosilicate glass at 1000 °C. J. Nucl. Mater. 452, 178–188 (2014).
McKeown, D. A., Muller, I. S. & Pegg, I. L. Iodine valence and local environments in borosilicate waste glasses using X-ray absorption spectroscopy. J. Nucl. Mater. 456, 182–191 (2015).
Muller, I. S., McKeown, D. A. & Pegg, I. L. Structural behavior of Tc and I ions in nuclear waste. Glass Procedia Mater. Sci. 7, 53–59 (2014).
Riley, B. J., Vienna, J. D., Strachan, D. M., McCloy, J. S. & Jerden, J. L. Materials and processes for the effective capture and immobilization of radioiodine: a review. J. Nucl. Mater. 470, 307–326 (2016).
Maddrell, E., Gandy, A. & Stennett, M. The durability of iodide sodalite. J. Nucl. Mater. 449, 168–172 (2014).
Du, L. S. & Stebbins, J. F. Network connectivity in aluminoborosilicate glasses: a high-resolution 11B, 27Al and 17O NMR study. J. Non Cryst. Solids 351, 3508–3520 (2005).
Smedskjaer, M. M. et al. Sodium diffusion in boroaluminosilicate glasses. J. Non Cryst. Solids 357, 3744–3750 (2011).
Le Losq, C., Neuville, D. R., Florian, P., Henderson, G. S. & Massiot, D. The role of Al3+ on rheology and structural changes in sodium silicate and aluminosilicate glasses and melts. Geochim. Cosmochim. Acta 126, 495–517 (2014).
Jolivet, V., Morizet, Y., Paris, M. & Suzuki-Muresan, T. High pressure experimental study on iodine solution mechanisms in nuclear waste glasses. J. Nucl. Mater. 533, 152112 (2020).
Jiřička, A., Vienna, J. D., Hrma, P. & Strachan, D. M. The effect of experimental conditions and evaluation techniques on the alteration of low activity glasses by vapor hydration. J. Non Cryst. Solids 292, 25–43 (2001).
Jegou, C., Narayanasamy, S. & Angeli, F. Short communication on the Influence of the temperature between 30 and 70 °C on the hydration of SON68 nuclear waste glass in a vapour phase. J. Nucl. Mater. 545, 152738 (2021).
Gast, J. A. & Thompson, T. G. Evaporation of boric acid from sea water. Tellus 11, 344–347 (1959).
Ishikawa, T. & Nakamura, E. Suppression of boron volatilization from a hydrofluoric acid solution using a boron-mannitol complex. Anal. Chem. 62, 2612–2616 (1990).
Goetschius, K. L., Beuerlein, M. A., Bischoff, C. M. & Brow, R. K. Dissolution behavior of ternary alkali-alkaline earth-borate glasses in water. J. Non Cryst. Solids 487, 12–18 (2018).
Zapol, P., He, H., Kwon, K. D. & Criscenti, L. J. First-principles study of hydrolysis reaction barriers in a sodium borosilicate glass. Int. J. Appl. Glas. Sci. 4, 395–407 (2013).
Gin, S. et al. The fate of silicon during glass corrosion under alkaline conditions: a mechanistic and kinetic study with the International Simple Glass. Geochim. Cosmochim. Acta 151, 68–85 (2015).
Neill, L. et al. Various effects of magnetite on international simple glass (ISG) dissolution: implications for the long-term durability of nuclear glasses. npj Mater. Degrad. 1, 1–11 (2017).
Ducasse, T. et al. Alteration of synthetic basaltic glass in silica saturated conditions: analogy with nuclear glass. Appl. Geochem. 97, 19–31 (2018).
Angeli, F., Gaillard, M., Jollivet, P. & Charpentier, T. Influence of glass composition and alteration solution on leached silicate glass structure: a solid-state NMR investigation. Geochim. Cosmochim. Acta 70, 2577–2590 (2006).
Kaspar, T. C. et al. Physical and optical properties of the International Simple. Glass npj Mater. Degrad. 3, 15 (2019).
Pitzer, K. S., Bradley, D. J., Rogers, P. S. Z. & Al., E. Thermodynamics of high temperature brines. In American Chemical Society-American Society for Testing and Materials Symposium (United States, 1979).
Efimov, A. M., Pogareva, V. G. & Shashkin, A. V. Water-related bands in the IR absorption spectra of silicate glasses. J. Non Cryst. Solids 332, 93–114 (2003).
Navarra, G., Iliopoulos, I., Militello, V., Rotolo, S. G. & Leone, M. OH-related infrared absorption bands in oxide glasses. J. Non Cryst. Solids 351, 1796–1800 (2005).
Acknowledgements
The authors acknowledge CEA and Valentin Jolivet from LPG-Nantes for supplying glass samples. We would like to thank the Région Pays de la Loire and the Pari Scientifique ‘CIPress’ project which provided financial support to this study. Special thanks to Nicolas Stéphant and Jean-Yves Mevellec from Institut des Matériaux de Nantes (IMN) for SEM analyses and FTIR analysis, respectively.
Author information
Authors and Affiliations
Contributions
H.Z. was responsible for the experimental analysis and for writing the paper. T.S., S.G., and A.A. supervised the study. Y.M. provided help for Raman data interpretation. All the authors helped with paper editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, H., Suzuki-Muresan, T., Morizet, Y. et al. Investigation on boron and iodine behavior during nuclear glass vapor hydration. npj Mater Degrad 5, 10 (2021). https://doi.org/10.1038/s41529-021-00157-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-021-00157-6
- Springer Nature Limited
This article is cited by
-
A review of glass corrosion: the unique contribution of studying ancient glass to validate glass alteration models
npj Materials Degradation (2023)
-
Borosilicate glass alteration in vapor phase and aqueous medium
npj Materials Degradation (2022)
-
Aqueous alteration of silicate glass: state of knowledge and perspectives
npj Materials Degradation (2021)