
Abstract
A data-driven framework is presented for building magneto-elastic machine-learning interatomic potentials (ML-IAPs) for large-scale spin-lattice dynamics simulations. The magneto-elastic ML-IAPs are constructed by coupling a collective atomic spin model with an ML-IAP. Together they represent a potential energy surface from which the mechanical forces on the atoms and the precession dynamics of the atomic spins are computed. Both the atomic spin model and the ML-IAP are parametrized on data from first-principles calculations. We demonstrate the efficacy of our data-driven framework across magneto-structural phase transitions by generating a magneto-elastic ML-IAP for α-iron. The combined potential energy surface yields excellent agreement with first-principles magneto-elastic calculations and quantitative predictions of diverse materials properties including bulk modulus, magnetization, and specific heat across the ferromagnetic–paramagnetic phase transition.
Similar content being viewed by others


Introduction
Magnetism strongly influences thermomechanical properties in a large variety of materials, such as single-element magnetic metals1,2, steels3, high-entropy alloys4,5, nuclear fuels such as uranium dioxide6, magnetic oxides7,8, and numerous other classes of functional materials9. Despite the critical role of magnetism in the aforementioned materials classes, modeling efforts to study the interplay between structural and magnetic properties have been notably lacking. Furthermore, there are unanswered scientific questions regarding the significance of magnetism in matter that is shock-compressed10,11 or exposed to strong electromagnetic fields such as in coherent lights sources12,13, pulsed power and high magnetic fields facilities14,15. Properties of interest include phase transitions, thermal stability of magnetic defects, magneto-mechanical couplings, but many of these subjects are challenging or prohibited by state of the art computational tools.
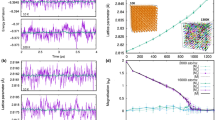
A prime but simple example of the computational advance made herein is the heat-capacity of α-iron displayed in Fig. 1. The experimental measurement of the heat capacity Cp diverges at the magnetic Curie transition, characteristic of a second-order phase transition16. Without a scalable coupled spin-lattice dynamics simulation environment, that properly accounts for thermal expansion and magnetic contribution to the pressure, reproducing the divergence of Cp (and of other thermomechanical properties) at the critical point is not possible.

The black triangles denote experimental measurements87,88, the red squares our simulation results, and black dashed line indicates the experimental Curie transition temperature. This illustrates the well-known ferromagnetic–paramagnetic phase transition, where the heat capacity diverges at the Curie temperature.
Accurate numerical simulations are critical for enabling technological advances, as they shape our fundamental understanding of the underlying solid state physics that dictates material behavior. Develo** high fidelity models, however, is challenging, because it necessitates capturing physical phenomena that occur across several length and time scales. This can only be achieved with sufficiently accurate multiscale simulation tools17,18, which is the focus of this work.
Classical molecular dynamics (MD) simulations19 provide a useful framework for multiscale modeling by leveraging interatomic potentials (IAPs) to represent the dynamics of atoms on a Born-Oppenheimer potential energy surface (PES)20. By utilizing massively parallel algorithms21 and long time-scale methodologies22, MD enables bridging first-principles with continuum-scale simulations23.
The absorption of machine learning (ML) techniques into the creation of interatomic potentials has lead to classical MD simulations that approach the accuracy of first-principles methods. A large number of these highly accurate ML-IAPs24,25,26,27,28,29,30 have been developed. In general, they are parameterized on training data (configuration energy, atomic forces) from first-principles methods like density functional theory (DFT)31 and utilize different flavors of ML model forms to construct the PES. While they have proven to be useful for large-scale simulations of materials properties32,33, further progress in multiscale modeling is hampered by the limitation of ML-IAPs to non-magnetic materials phenomena. Even with highly accurate ML-IAPs, state-of-the-art MD simulations cannot reproduce the divergent behavior of Cp near the critical point (Fig. 1) because they fail to account for the magnetic degrees of freedom34.
Coupling atomic spin dynamics with classical MD has been pioneered by Ma et al.35,36,37. Herein, a classical magnetic spin is assigned to each atom in addition to its position leading to a 6N-dimensional PES (5N if the magnetic spin norms are fixed), instead of the common 3N-dimensional PES in classical MD:
where rij = ri − rj denotes the relative position between atoms i and j, si the classical spin assigned to atom i, and N the number of atoms in the system. In most classical spin-lattice calculations, the 6N-dimensional PES is constructed by introducing an atomic spin model on top of a mechanical IAP35. For example, a common approach is to combine a distance-dependent Heisenberg Hamiltonian with an embedded-atom-method (EAM) potential37,38.
While these prior approaches recover experimental properties on a qualitative level39,40, their combined representation of phononic and magnetic degrees of freedom is not sufficiently consistent for providing quantitative predictions at the level of first-principles results. More recently, Ma et al. developed a magneto-elastic IAP for magnetic iron based on data from first-principles calculations41. However, this remained an isolated attempt as there is no general methodology for generating a magneto-elastic PES in a classical context that enables large-scale spin-lattice dynamics simulations for any magnetic material.
In this work, we overcome this methodological obstacle by providing a data-driven framework for generating magneto-elastic ML-IAPs that (1) provide a consistent representation of both mechanical and magnetic degrees of freedom and (2) achieve near first-principles accuracy. We refer to our new class of IAPs as magneto-elastic ML-IAPs as they generate a consistent PES accurately representing the magnetic degrees of freedom and the interplay between magnetic and elastic phenomena. Our framework couples an atomic spin model (Heisenberg Hamiltonian) with an ML-IAP and provides a unified magneto-elastic PES which yields the correct mechanical forces on the atoms in the MD framework. The Heisenberg Hamiltonian is parameterized with data from DFT spin-spiral calculations at different degrees of lattice compression. In constructing the ML-IAP, we leverage the flexible and data-driven spectral neighbor analysis potential (SNAP) methodology30 which is trained on a database of magnetic configurations generated using DFT calculations. We highlight the influence of magnetization dynamics on thermo-mechanical properties by assessing three different thermodynamic equilibration conditions. This allows us to conclude that a correction to the magnetization dynamics (achieved by adapting the temperature-rescaling method42) is necessary for accurate elastic predictions.
We apply our framework to generate a magneto-elastic ML-IAP for the α phase of iron. We demonstrate that our potential is transferable to an extended area of the phase diagram, corresponding to a temperature and pressure range of 0 to 1200 K and 0 to 13 GPa (up to the α → γ and α → ϵ transitions, respectively). The Curie temperature, which experimentally occurs at ~1045 K, lies within this parameter space. After presenting our training workflow, the “Results” section will probe the accuracy of our magneto-elastic ML-IAP by performing magneto-static comparisons to first-principles measurements. We then stress that our generated magneto-elastic ML-IAP can also be directly used in the LAMMPS package21 to perform magneto-dynamic simulations that take into account both the thermal expansion of the lattice and magnetic pressure due to spin disorder. This enables us to maintain a constant ambient pressure throughout all calculations of thermomechanical properties, consistent with conditions prevalent in experiments. As illustrated in Fig. 1, our framework allows us to perform pressure-controlled quantitative prediction of the critical behavior across a second-order phase transition within a classical spin-lattice dynamics simulation.
Results
In this section we outline our advancements in magnetic materials modeling. We first present our training workflow and subsequently assess our results by comparing both static and dynamic properties in α-iron against first-principles calculations and experiments.
Figure 2 displays our training workflow. Further details to each box in this diagram are presented as a subsection in the “Methods” section. All atomic configurations in the training set result from first-principles calculations performed with the same DFT setup (same pseudo-potential and energy cutoff, similar k-point densities) as detailed in the “Methods” section. In contrast to traditional force-matching approaches in the development of classical IAPs, we treat the magnetic and phononic degrees of freedom in the PES in a consistent and unified manner, as indicated by the exchange of information between spin Hamiltonian and SNAP potential parametrization steps. After parameterizing our atomic spin Hamiltonian by leveraging DFT spin-spiral results, its energy, forces, and stress contributions are subtracted from each atomic configuration in the first-principles training set. The ML-IAP is then trained to reproduce the non-magnetic component of the first-principles data. Finally, both components of the magneto-elastic PES are recombined to construct a unified magneto-elastic ML-IAP that is consistently trained on first-principles data. Optimization is handled by the DAKOTA software package43 in both fitting steps. For the SNAP component of the potential, DAKOTA optimizes the radial cutoff of the interaction along with the weights of each training data set (energy and force weights) to generate different candidate potentials. Those candidates potentials are then recombined with the spin Hamiltonian and tested against selected objective functions (mean-absolute errors (MAEs) in lattice constants, cohesive energies, elastic constants, forces and total energies). Table 1 summarizes the different groups of training data, the optimal weights obtained for each of those groups, and the corresponding energy and force MAEs. The target values for the objective functions are based on both experimental and DFT data, as outlined in Table 2. Objective function evaluations are done within LAMMPS21.

A training set of DFT calculations is partitioned into those that train the SNAP interatomic potential and those that train the spin Hamiltonian, respectively. A non-magnetic interatomic potential is fit to configuration energies and atomic forces after the spin Hamiltonian contribution is subtracted and is validated against magneto-elastic properties computed in LAMMPS. Optimization of the spin Hamiltonian and interatomic potential parameters is handled by DAKOTA.
Herein, the critical innovation that enables a leap forward in predictive simulations of magnetic materials is this data-driven workflow. Magnetic and phononic contributions to the PES are taken into account explicitly and any miscounting is avoided (for example, no double counting of the magnetic energy or contribution to the pressure). The obtained magneto-elastic ML-IAP can directly be used to run spin-lattice calculations in LAMMPS21,38,44.
Magneto-static accuracy
We first assess the quantitative agreement of our magneto-elastic ML-IAP by comparing with DFT results where magnetic order and elastic deformations are coupled. This is done by leveraging a particular subset of spin configurations referred to as spin-spirals, for which the energy and corresponding pressure can be evaluated from both DFT and classical magneto-elastic potential calculations. Details about definition and computation of spin-spirals can be found in the “Methods” section. Equation-of-state calculations (energy and pressure versus volume) are performed at the Γ point (corresponding to the purely ferromagnetic state) and for spin-spirals corresponding to q-vectors along the ΓH and ΓP high-symmetry lines. The calculations at the Γ point represent the magnetic ground state and, hence, serve as a point of reference for the spin spiral calculations. The geometric orientation of the various computed spin spirals is visualized in Fig. 3. The first set (q = 0.01 along ΓH and q = 0.07 along ΓP) represents long spirals, close to the Γ point, the second set (q = 0.1 along ΓH and q = 0.14 in ΓP) represents spirals with intermediate periodicity, and the last set (q = 0.2 along ΓH and q = 0.21 along ΓP) is chosen close to the borders of the magnetic training set (see red demarcation lines in Fig. 5 in the “Methods” section). The DFT results are obtained by leveraging the generalized Bloch theorem, whereas our classical spin-lattice calculations were performed by generating the corresponding supercells (details given in the “Methods” section).

Plots of the equation of state data from first-principles calculations (VASP computations) and our magneto-elastic ML-IAP (LAMMPS computations) for seven different spin-spirals: a Γ point (b) vectors along the ΓH high-symmetry line, and c vectors along the ΓP high-symmetry line. Visualizations of the corresponding spin-spiral supercells and associated q-vectors are shown to the right of and above each plot, respectively.
Excellent agreement between our classical spin-lattice model and DFT is achieved at the Γ point and for the two first q-vectors on each high-symmetry line (q = 0.01 and q = 0.1 along ΓH, q = 0.07 and q = 0.14 along ΓP) in the pressure range relevant for the α-phase of iron (up to 13 GPa which corresponds to the α → ϵ transition). At higher q-vector values, the energy and pressure predictions of our atomic spin-lattice model still agree reasonably well with the DFT calculations. The observed deviation from the DFT results can be explained by the limitations of our atomic spin-lattice model: as both the pressure and the relative angle between neighboring spins increase, fluctuations of the atomic spin norms become more important. As discussed in the “Methods” and “Discussion” sections, these are not included in the Hamiltonian of our atomic spin-lattice model.
Magneto-dynamic accuracy
Turning now to spin-lattice dynamics calculations based on our magneto-elastic ML-IAP (as detailed in the “Methods” section), we assess the quantitative accuracy with respect to experimental measurements of changes in magnetic and thermoelastic properties as the material is heated. In making this comparison, it is necessary to choose which thermodynamic state variables will be held fixed and which will be allowed to vary with temperature. Spin-lattice dynamics algorithms have been developed for simulations in a canonical ensemble which preserves the number of particles, the volume, and the temperature in the system37. Our first set of simulation conditions, referred to as fixed-volume conditions (FVC), hold the volume fixed while running dynamics in the canonical ensemble at specified values of the lattice and spin temperatures. A disadvantage of this choice is that the pressure steadily increases as heat is added to the material, in contradiction to the experimental observations, which are conducted at constant pressure. To this date, an isobaric spin-lattice algorithm has not been developed (preserving the system’s pressure rather than its volume). However, our methodology as implemented in LAMMPS enables us to compute the magnetic contribution to the pressure. By alternating thermalization (coupled spin-lattice dynamics in a canonical ensemble) and pressure equilibration (frozen spin configuration in an isobaric ensemble) steps, it is possible to control the pressure of our spin-lattice system. Hence, we refer to calculations performed in this pressure-controlled canonical ensemble as pressure-controlled conditions (PCC). In both conditions, the temperature of the spin and lattice subsystems is set using two separate Langevin thermostats (one acting on the spins, the other on the lattice)37. Finally, this enables us to define a third set of conditions: in addition to controlling the pressure, the spin thermostat can be set to match a given magnetization value (i.e., the experimental magnetization) rather than a temperature. We refer to this as pressure-controlled and magnetization-controlled conditions (PCMCC). Figure 6 in the “Methods” section displays the different definitions of the spin temperature and the evolution of the pressure for those three different conditions.
In practice, FVC, PCC, and PCMCC only differ in their equilibration conditions (control of pressure and/or magnetization), as each of the corresponding simulations are performed in a canonical ensemble. We illustrate the predictive capability of our magneto-elastic ML-IAP in α-iron for these equilibration conditions in Fig. 4a–f (FVC:  , PCC:
, PCC:  , PCMCC:
, PCMCC:  ). The agreement of the following magneto-elastic properties with experimental results is assessed: magnetization (Fig. 4a), heat-capacity Cp (Fig. 4b), thermal expansion (cell volume on Fig. 4c), bulk modulus (Fig. 4d), and two shear constants, (c11 − c12)/2 and c44 (Fig. 4e, f). The “Spin-lattice dynamics” subsection of the Methods section details the computation of those temperature-dependent elastic constants.
). The agreement of the following magneto-elastic properties with experimental results is assessed: magnetization (Fig. 4a), heat-capacity Cp (Fig. 4b), thermal expansion (cell volume on Fig. 4c), bulk modulus (Fig. 4d), and two shear constants, (c11 − c12)/2 and c44 (Fig. 4e, f). The “Spin-lattice dynamics” subsection of the Methods section details the computation of those temperature-dependent elastic constants.

Plots a–f show magnetoelastic data obtained with our magneto-elastic ML-IAP. The green ( ), blue (
), blue ( ), and red (
), and red ( ) markers indicate the choice of equilibration conditions: fixed-volume conditions (FVC), pressure-controlled conditions (PCC) and pressure-controlled and magnetization-controlled conditions (PCMCC), respectively. In all plots, experimental data (extracted from five different references87,88,89,90,91) is denoted by the filled triangles (▲), and the dotted black lines (
) markers indicate the choice of equilibration conditions: fixed-volume conditions (FVC), pressure-controlled conditions (PCC) and pressure-controlled and magnetization-controlled conditions (PCMCC), respectively. In all plots, experimental data (extracted from five different references87,88,89,90,91) is denoted by the filled triangles (▲), and the dotted black lines ( ) represent the experimental Curie temperature. The plots in a, b show magnetization and specific heat comparisons between different ensembles and experiments. The light blue region in (b) indicates the low temperature regime T ≲ 250 K where quantum effects reduce the experimental heat capacity below the classical Dulong-Petit limiting value of 3R45. The data in plot c illustrates how the lattice expands with temperature. An inherent offset exists between our model (trained to match the DFT data at 0 K) and experimental measurements. Plots d–f show d bulk modulus, e (c11 − c12)/2 shear constant, and f c44 shear constant for the three aforementioned sets of conditions.
) represent the experimental Curie temperature. The plots in a, b show magnetization and specific heat comparisons between different ensembles and experiments. The light blue region in (b) indicates the low temperature regime T ≲ 250 K where quantum effects reduce the experimental heat capacity below the classical Dulong-Petit limiting value of 3R45. The data in plot c illustrates how the lattice expands with temperature. An inherent offset exists between our model (trained to match the DFT data at 0 K) and experimental measurements. Plots d–f show d bulk modulus, e (c11 − c12)/2 shear constant, and f c44 shear constant for the three aforementioned sets of conditions.
We first work under the FVC ( ), kee** a constant volume and equal spin and lattice temperatures (Figs. 4c and 6). At constant volume, our model predicts a Curie temperature of ~716 K (Fig. 4a). Specific heat calculations shown in Fig. 4b were performed by computing the derivative of the internal energy, taking both the lattice and magnetic contributions into account. The SNAP contribution (lattice only) was first isolated and determined to be a constant value of 26.4 Jmol−1 K−1, in good agreement with the Dulong-Petit value of 3R45. The magnetic contribution offsets the total specific heat at low temperature, as the magnetization steadily decreases (thus steadily increasing the magnetic energy). Also at low temperature, deviation between simulations and experiment (highlighted by the semi-transparent blue region in Fig. 4b) occurs due to quantum effects which reduce the experimental heat capacity below the 3R value. The FVC heat-capacity is determined at constant volume, although we use the symbol Cp on the axis label because the enhanced simulations described below are indeed conducted at constant pressure conditions. In those constant volume conditions, the pressure evolution with temperature increase is substantial (up to 12 GPa, almost corresponding to the α → ϵ transition, as can be seen on Fig. 6), which has a strong impact on the underlying elastic properties. Interestingly, at the Curie temperature (here 716 K), the increasing pressure exhibits an inflection point, confirming the importance of spin fluctuations on the thermoelastic properties. The temperature dependence of three elastic constants is shown in Fig. 4d–f. For the bulk modulus, FVC does not agree well with experimental data, especially at higher temperatures. The FVC results tend to overestimate the stiffness, which most likely arises from the build-up of thermal stresses in the material. Under these conditions a nearly temperature-invariant c44 response is predicted, which is in strong contrast to trends in experiment. Despite these shortcomings, the FVC calculations actually match the experimental data for shear constant (c11 − c12)/2 relatively well throughout the entire temperature range. In general, the fixed volume assumption made under FVC fails to account for thermal expansion, leading to incorrect elastic predictions.
), kee** a constant volume and equal spin and lattice temperatures (Figs. 4c and 6). At constant volume, our model predicts a Curie temperature of ~716 K (Fig. 4a). Specific heat calculations shown in Fig. 4b were performed by computing the derivative of the internal energy, taking both the lattice and magnetic contributions into account. The SNAP contribution (lattice only) was first isolated and determined to be a constant value of 26.4 Jmol−1 K−1, in good agreement with the Dulong-Petit value of 3R45. The magnetic contribution offsets the total specific heat at low temperature, as the magnetization steadily decreases (thus steadily increasing the magnetic energy). Also at low temperature, deviation between simulations and experiment (highlighted by the semi-transparent blue region in Fig. 4b) occurs due to quantum effects which reduce the experimental heat capacity below the 3R value. The FVC heat-capacity is determined at constant volume, although we use the symbol Cp on the axis label because the enhanced simulations described below are indeed conducted at constant pressure conditions. In those constant volume conditions, the pressure evolution with temperature increase is substantial (up to 12 GPa, almost corresponding to the α → ϵ transition, as can be seen on Fig. 6), which has a strong impact on the underlying elastic properties. Interestingly, at the Curie temperature (here 716 K), the increasing pressure exhibits an inflection point, confirming the importance of spin fluctuations on the thermoelastic properties. The temperature dependence of three elastic constants is shown in Fig. 4d–f. For the bulk modulus, FVC does not agree well with experimental data, especially at higher temperatures. The FVC results tend to overestimate the stiffness, which most likely arises from the build-up of thermal stresses in the material. Under these conditions a nearly temperature-invariant c44 response is predicted, which is in strong contrast to trends in experiment. Despite these shortcomings, the FVC calculations actually match the experimental data for shear constant (c11 − c12)/2 relatively well throughout the entire temperature range. In general, the fixed volume assumption made under FVC fails to account for thermal expansion, leading to incorrect elastic predictions.
We correct this shortcoming of the model by working under PCC ( ) which allows for thermal expansion. As can be seen on Fig. 4c, the cell volumes are relaxed at each finite temperature, until the pressure in the system drops to ~0 GPa. As shown in Fig. 4a, the thermal expansion incorrectly moves the onset of Curie transition to ~536 K. As the average interatomic distance increases, the strength of the exchange interaction is lowered, thus decreasing the transition temperature. The computed heat-capacity (Fig. 4b) now corresponds to the derivative of the free energy, and to an actual Cp measurement. However, as in the FVC, the low agreement between the experimental and computed magnetization evolution leads to an offset in the initial Cp and does not match the Dulong-Petit value at low temperature. The PCC fares better in reproducing the experimental bulk modulus up to the Curie transition (no hardening observed). PCC also does better in terms of the shear constant c44, as it is able to reproduce the thermal softening seen in experiments. However, for shear constant (c11 − c12)/2, PCC underestimates the extent of the thermal softening. Overall, PCC does better than FVC in terms of elastic properties, but deviates more in terms of magnetic predictions compared to experiment. By shifting the Curie transition towards lower temperatures, it reduces the range of validity of our elastic calculations.
) which allows for thermal expansion. As can be seen on Fig. 4c, the cell volumes are relaxed at each finite temperature, until the pressure in the system drops to ~0 GPa. As shown in Fig. 4a, the thermal expansion incorrectly moves the onset of Curie transition to ~536 K. As the average interatomic distance increases, the strength of the exchange interaction is lowered, thus decreasing the transition temperature. The computed heat-capacity (Fig. 4b) now corresponds to the derivative of the free energy, and to an actual Cp measurement. However, as in the FVC, the low agreement between the experimental and computed magnetization evolution leads to an offset in the initial Cp and does not match the Dulong-Petit value at low temperature. The PCC fares better in reproducing the experimental bulk modulus up to the Curie transition (no hardening observed). PCC also does better in terms of the shear constant c44, as it is able to reproduce the thermal softening seen in experiments. However, for shear constant (c11 − c12)/2, PCC underestimates the extent of the thermal softening. Overall, PCC does better than FVC in terms of elastic properties, but deviates more in terms of magnetic predictions compared to experiment. By shifting the Curie transition towards lower temperatures, it reduces the range of validity of our elastic calculations.
In order to improve the magnetic predictions of α-iron, we finally consider the PCMCC scheme ( ). The present calculations are aimed at probing the influence of the magnetization dynamics on thermo-mechanical properties of materials. For FVC and PCC, two main physical limitations associated to the classical spin dynamics model can be observed: an offset of the predicted Curie transition, and the magnetization versus temperature trend (as clearly displayed on Fig. 4a, b). The Curie transition offset is mainly attributed to our choice of parametrization of the magnetic interactions (see “Methods” section). Compared to experimental measurements, our low temperature magnetization trend is immediately decreasing. This, for example, strongly impacts the Cp measurements (Fig. 4b), as it generates a large magnetic energy decrease, leading to an offset from the Dulong-Petit value at low temperature. In order to improve this limitation of classical spin dynamics and better observe the influence of magnetization dynamics on the thermo-mechanical properties predicted by our model, we follow the approach developed by Evans et al.42. However, our simulations account for thermal expansion, so that our magnetic temperature rescaling has to be slightly modified compared to their approach. A full magnetization versus temperature calculation in the PCC has to be performed before evaluating which spin temperature corresponds to a give lattice temperature (see “Methods” section for more details). Figure 4a shows that the obtained magnetization under PCMCC closely matches that of experiment. Most prominently, the resulting Cp agrees well with experiments (Fig. 4b). The Dulong-Petit value is recovered at low temperature, and the Cp discontinuity at the Curie transition is well captured. The thermal expansion trend is also in much better agreement with experiments, with very comparable slopes between ~200 and 750 K (Fig. 4c). Up to ~600 K, PCMCC agree very well with the experimental values for (c11 − c12)/2 (Fig. 4e) but at 800–1000 K a slight hardening is observed, which contradicts experimental data. For the bulk modulus, PCMCC correctly predicts the nearly linear trend up to the Curie temperature.
). The present calculations are aimed at probing the influence of the magnetization dynamics on thermo-mechanical properties of materials. For FVC and PCC, two main physical limitations associated to the classical spin dynamics model can be observed: an offset of the predicted Curie transition, and the magnetization versus temperature trend (as clearly displayed on Fig. 4a, b). The Curie transition offset is mainly attributed to our choice of parametrization of the magnetic interactions (see “Methods” section). Compared to experimental measurements, our low temperature magnetization trend is immediately decreasing. This, for example, strongly impacts the Cp measurements (Fig. 4b), as it generates a large magnetic energy decrease, leading to an offset from the Dulong-Petit value at low temperature. In order to improve this limitation of classical spin dynamics and better observe the influence of magnetization dynamics on the thermo-mechanical properties predicted by our model, we follow the approach developed by Evans et al.42. However, our simulations account for thermal expansion, so that our magnetic temperature rescaling has to be slightly modified compared to their approach. A full magnetization versus temperature calculation in the PCC has to be performed before evaluating which spin temperature corresponds to a give lattice temperature (see “Methods” section for more details). Figure 4a shows that the obtained magnetization under PCMCC closely matches that of experiment. Most prominently, the resulting Cp agrees well with experiments (Fig. 4b). The Dulong-Petit value is recovered at low temperature, and the Cp discontinuity at the Curie transition is well captured. The thermal expansion trend is also in much better agreement with experiments, with very comparable slopes between ~200 and 750 K (Fig. 4c). Up to ~600 K, PCMCC agree very well with the experimental values for (c11 − c12)/2 (Fig. 4e) but at 800–1000 K a slight hardening is observed, which contradicts experimental data. For the bulk modulus, PCMCC correctly predicts the nearly linear trend up to the Curie temperature.
We note that in all three sets of conditions, a rapid increase of about 25–30 GPa in the bulk modulus is observed as we move across the critical point. This jump was found to be strongly impacted by the underlying mechanical potential. The prediction accuracy could possibly be improved by including additional, finite-temperature objective functions in the fitting procedure. The PCMCC prediction of the shear constant c44 closely matches the PCC data. This tends to indicate that this shear constant c44 is not impacted significantly by the spin dynamics. For both pressure controlled conditions (PCC and PCMCC) the maximum deviation from experiments occurs near 700 K and is ~14%.
Discussion
We presented a data-driven framework for automated generation of magneto-elastic ML-IAPs which enable large-scale spin-lattice dynamics simulations for any magnetic material in LAMMPS. This framework was demonstrated by generating a robust magneto-elastic ML-IAP for α-iron. First we investigated the magneto-static accuracy (energy and pressure) with respect to equivalent first-principles calculations. It was demonstrated that the generated magneto-elastic ML-IAP (which represents the corresponding 5-N dimensional PES) is in close agreement with first-principles magneto-elastic calculations. This was achieved by properly partitioning the PES into magnetic and mechanical degrees of freedom. Subsequently, we investigated the magneto-dynamic accuracy by comparing predicted finite temperature magneto-elastic properties (magnetization, heat-capacity, thermal-expansion, bulk modulus, and shear constants) across the ferromagnetic–paramagnetic phase transition from spin-lattice dynamics simulations against data from experiments. In the course of this, we analyzed the choice of simulation conditions (control of pressure and magnetization) and highlighted the importance of thermal and magnetic pressure contributions. This is an important advance over traditional classical magnetization dynamics methods, where contributions from thermal expansion or spin pressure due to disorder are negated. We demonstrated that spin-lattice dynamics simulations of controlled pressure and constrained magnetization yields qualitative agreement with the measured magneto-elastic properties.
Our framework enables predictions of critical properties across the second-order phase transition within classical spin-lattice dynamics simulations, such as the divergent behavior of the heat capacity around the Curie temperature (Figs. 1 and 4b). We provide a more comprehensive perspective on our results by comparing them within the context of other first-principles and classical methods. At low temperature, first-principles methods can capture the electronic component of the heat-capacity, up to the Dulong-Petit value45,46 (the difference with our model is highlighted by the blue area on Fig. 4b). However, computing Cp across the Curie transition requires a dynamic treatment of large spin-ensembles whose calculation is computationally expensive in terms of first-principles methods. Classical IAPs do not explicitly treat magnetic degrees of freedom and, thus, cannot reproduce the effects of this magnetic phase-transition47. An empirical model which is based on first-principles calculations and accounts for electronic, phononic and magnetic degrees of freedom gave excellent agreement with the experimental Cp curve of α-iron up to the Curie temperature48. However, this model does not extend above the Curie temperature, does not account for the pressure generated by the corresponding spin configurations, and cannot be easily extended to other thermomechanical properties. Thus, for a range of temperature from about 250 to 1200 K, our model provides with a set of very good predictions, obtained for the computational cost of classical MD calculations only.
We conclude the discussion of our results by pointing out limitations of the present method and future prospects. First, note that the agreement to the experimental Curie transition (Tc ≈ 716 K in a fixed volume calculation) could have been adjusted by parameterizing the spin potential on a smaller range of the high-symmetry lines (see Fig. 5), or by adding an objective function aimed at matching the experimental value in the spin-potential fitting procedure. However, this additional constraint would have worsened the agreement of our model with the DFT energy and pressure results (as displayed on Fig. 3) and would contradict the overall objective of this work.

Comparison of spin-spiral results along sections of the ΓH and ΓP high-symmetry lines. The upper plot displays the per-atom energy, the middle one the atomic moment fluctuations (in Bohr magneton per atom), and on the bottom the evolution of the pressure. The energy and pressure fluctuations are plotted with respect to the magnetic ground state at the Γ point. The green and red dots represent experimental measurements obtained by Loong et al.92 and Lynn93. In all three plots, the dashed lines correspond to the DFT results, and the continuous lines to our classical model results, whereas the line color (black or blue) corresponds to the lattice compression (0% or 2%, respectively). In the middle plot, the green dashed horizontal line represent the experimental equilibrium value (2.2μB per atom), which is the constant value chosen in our model. In all three plots, the red vertical dashed lines are delimiting the q-vectors on which our spin Hamiltonian was parametrized.
For temperatures below ~250 K, our classical framework cannot access the quantized free energy, and is thus unable to accurately reproduce the trends of all the quantities being its derivatives (Cp, elastic constants, ...). This is reducing the agreement versus experiments of the magneto-dynamic accuracy measurements displayed on Fig. 4 at low temperature, and can be seen as a limitation of our classical approach49.
Another limitation of our work lies in the simplicity of the spin Hamiltonian model used. The Heinsenberg Hamiltonian (as well as its extended forms) is a robust model that can be transposed to a large number of magnetic systems and applications7,39,50,51. However, finding a unique lattice dependence (herein the Bethe-Slater function) and its’ associated accurate parametrization is very challenging. Comparing Fig. 4a and Fig. 5 reveals some of this difficulty: although the predicted spin-spiral energies are in good agreement with the DFT calculations, the corresponding pressure dependence and the Curie temperature predictions (in the FVC and PCC) are departing from their expected values. Other parametrizations of the spin Hamiltonian could have been performed, either improving the Curie temperature predictions, or the generated magnetic pressure, but worsening the comparison versus DFT of the spin-spiral energies. In this study, we decided to focus our fit on the spin-spiral energies for two main reasons:
-
(i)
Our approach aims at develo** magneto-elastic ML-IAPs trained on first-principles data without double counting of the magnetic component of the PES. Thus, obtaining an energetic dependence of spin configurations close to the DFT results should remain the priority.
-
(ii)
The approximations of the model could be better controlled this way. Indeed, the spin model we used was developed to reproduce magnon energetics, and does not account for fluctuations of the spin norms (although this is not a limitation of the presented framework). Thus, a stronger emphasis was set on reproducing the spin-spiral energies for which the spin norm fluctuations remain under 5% fluctuations from the Γ point value (as displayed on Fig. 5 in the “Methods” section). Different forms of spin Hamiltonians, such as spin-cluster expansions, might be a promising route to improving the accuracy of the magnetic component of the PES by both accounting for the fluctuation of the magnetic moment magnitudes and many-body spin interactions52,53. A straightforward extension of this work could combine recently developed extended spin Hamiltonians with first-principles studies, and apply our formalism to extend our α-iron magneto-elastic ML-IAP to account for defect configurations54,55, Cr clustering56,57, and magneto-structural phase-transitions11,58.
Our study emphasizes the important influence of magnetization dynamics on thermo-mechanical properties, even for a simple ferromagnet such as iron where the magneto-elasticity is rather weak. We showed that the well-known departures from the experimental magnetization curve observed in classical spin dynamics have a strong influence on the predictions of the model. In this work, we chose to follow the approach proposed by Evans et al.42 to correct this shortcoming of the approach. This allowed us to accurately represent the temperature influence on thermo-mechanical properties of iron through its α-phase and across the Curie transition. Enhanced magnetic thermostats have been proposed in order to better match the experimental magnetic transition versus temperature59,60. Such thermostats could be implemented in LAMMPS and used to replace the magnetization-controlled conditions defined in the “Results” section. This could extend the range validity of our framework to areas of phase-diagrams where the magnetization distribution is not well measured (for example in the ϵ phase of iron).
A recent study added a magnetic contribution to the set of descriptors used in a moment-tensor ML-IAP61. Although this approach does not explicitly simulate the magnetization dynamics (and its effects on thermomechanical properties), the authors demonstrated remarkable improvement in terms of error convergence. At this stage of our work, we believe improving the modeling of the magnetic component of the PES remains our first priority (and thus implementing and fitting improved spin Hamiltonians, as discussed above). However, depending on the success of this first effort, this complementary approach could be leveraged to improve the accuracy of our magneto-elastic ML-IAPs.
In summary, we have presented a new computational framework for simulations of magneto-elastic materials properties near first-principles accuracy. By leveraging the flexibility of ML-IAPs, our data-driven workflow enables to model the interplay between magnetic and phononic dynamics for a large class of magnetic materials. Furthermore, our straightforward connection to the LAMMPS package makes it possible to perform large-scale quantitative magneto-elastic predictions over controlled pressure and temperature spaces, hitherto study unexplored magneto-dynamics properties of materials.
Methods
Density functional theory calculations
Parameterizing both the ML-IAP and the magnetic Heisenberg Hamiltonian relies on data computed using spin-dependent DFT calculations. They were performed using VASP62,63. In all calculations the PBE64 exchange-correlation functional was employed. We used PAW pseudopotentials65 with 8 valence electrons and a core radius of rc = 2.3aB. The plane wave cutoff was set to 320 eV and the convergence in each self-consistency cycle was set to 10−8. The Fermi-Dirac smearing scheme with a width of 0.026 eV was used. The Brillouin zone was sampled on a 10 × 10 × 10 grid of k-points. The number of bands used was 224 per atom.
Spin-spiral calculations
Spin-spirals define a subset of non-collinear magnetic states. In this work, we leverage spin-spirals as a convenient tool to perform one-to-one comparisons between first-principles and classical magneto-elastic calculations. They can be defined as follows:
where q is the spin-spiral vector, R0j is the position of atom j relative to a central atom 0, sj is the spin on atom j, and θ is a constant angle between the spins and the spin-spiral vector (often referred to as cone angle)51. \({{{\hat{\boldsymbol{x}}}}}\), \({{{\hat{\boldsymbol{y}}}}}\), and \({{{\hat{\boldsymbol{z}}}}}\) are the unit vectors along [100], [010], and [001], respectively. Our calculations are restricted to θ = π/2, corresponding to flat spin-spirals in the (001) plane.
First-principles calculations of the per-atom energy and the pressure corresponding to spin-spiral states are performed using DFT by leveraging the frozen-magnon approach66,67 and the generalized Bloch theorem68 as implemented in VASP69. We consider a primitive cell of one atom. A 10 × 10 × 10 k-point grid, an energy cutoff of 320 eV, and 224 bands proved sufficient to reach the level of accuracy expected in our model (as can be seen in Fig. 5).
Classical calculations are performed by using Eq. (2) to generate supercells accommodating the spin-spirals corresponding to the q-vectors used in the DFT calculations. Based on a given supercell and a spin Hamiltonian, the per-atom energy and pressure are computed using the SPIN package of LAMMPS21,38.
Spin Hamiltonian
A spin Hamiltonian is used to model the energy, mechanical forces, and pressure contributions of magnetic configurations. Rosengaard and Johansson50 and Szilva et al.70 showed that adding a biquadratic term to the classical Heisenberg Hamiltonian improves the accuracy of magnetic excitations in 3-d transition ferromagnets. We adopted their Hamiltonian form:
where si and sj are classical atomic spins of unit length located on atoms i and j, \(J\left({r}_{ij}\right)\) and \(K\left({r}_{ij}\right)\) (in eV) are magnetic exchange functions, and rij is the interatomic distance between magnetic atoms i and j. The two terms in Eq. (3) are offset by subtracting the spin ground state (corresponding to a purely ferromagnetic situation), as detailed in Ma et al.35. Although this offset of the exchange energy does not affect the precession dynamics of the spins, it allows to offset the corresponding mechanical forces. Without this additional term, the magnetic contribution to the forces and the pressure are not zero at the energy ground state. For the exchange interaction terms \(J\left({r}_{ij}\right)\,\) and \(K\left({r}_{ij}\right)\), the interatomic dependence is taken into account through the following function based on an approximation of the Bethe-Slater curve71,72:
where α denotes the interaction energy, δ the interaction decay length, γ a dimensionless curvature parameter, r = rij is the radial distance between atoms i and j, and \({{\Theta }}\left({R}_{c}-r\right)\) a Heaviside step function for the radial cutoff Rc. This assumes that the interaction decays rapidly with the interatomic distance, consistent with former calculations70,73. We set Rc = 5 Å to include five neighbor shells, as Pajda et al.73 showed that the exchange interaction decays slower along the [111] direction in α-iron.
Using Eq. (3) and leveraging the generalized spin-lattice Poisson bracket as defined by Yang et al.74, the magnetic precession vectors (ωi), mechanical forces (Fi), and their corresponding virial components (\(W\left({{{{\boldsymbol{r}}}}}^{N}\right)\)) are derived:
where rN denotes a 3N size vector of all atomic positions and ri the position vector of atom i. The primed sum in the above expression for the virial indicates that force contributions on atoms that are periodic images must be summed separately75. The total pressure is obtained by combining this virial with thermal and mechanical contributions. The precession vectors (ωi) are defined following the definitions of Evans76. In Eq. (5), γ is the gyromagnetic ratio (γ ≈ 0.176 rad ⋅ THz ⋅ T−1), and μi is the atomic spin norm, in Bohr magneton. This yields a precession frequency in rad ⋅ THz (corresponding to the metal units of LAMMPS).
The spin Hamiltonian is used to reproduce spin-spiral energy and pressure reference results obtained from DFT. They are sampled along two high-symmetry lines, ΓH and ΓP, and for two different lattice constant values (corresponding to the equilibrium bulk value and to a lattice compression of 2%). This allows us to encapsulate in the model the influence of lattice compression on the spin stiffness and the Curie temperature, which was experimentally and theoretically predicted to be small77,78,79. Figure 5 displays the excellent agreement obtained between our first-principles spin-spiral energies and experimental measurements.
Our current spin Hamiltonian does not account for fluctuations of the magnetic moment magnitudes, i.e., the norm of atomic spins remains constant in our calculations. As can be seen in Fig. 5, this is not the case for our DFT results, as those fluctuations can become important when departing from the Γ point. We thus decided to parameterize our model only on spin-spirals corresponding to q-vectors for which the spin norm deviates from the ferromagnetic value (≈2.2μB/atom at the Γ point) by <5%. The red dashed lines in Fig. 5 delimit this q-vector range.
Finally, we used the single objective genetic algorithm within the DAKOTA software package43 to optimize the six coefficients of \(J\left({r}_{ij}\right)\) and \(K\left({r}_{ij}\right)\) in order to obtain the best possible agreement between our reference DFT spin-spiral energy and pressure results and our spin model. Figure 5 displays the obtained result. As can be seen in Fig. 4, for a fixed-volume calculation, our spin Hamiltonian predicts a Curie temperature of 716 K. Note that a better match of the DFT spin-spiral energies would yield a larger spin-stiffness, and thus a better agreement for the Curie temperature. However, this would worsen the pressure agreement.
Spin–orbit coupling effects were included by accounting for an iron-type cubic anisotropy80:
with K1 = 0.001 eV and \({K}_{2}^{(c)}=0.0005\) eV the intensity coefficients corresponding to α-iron. The cubic anisotropy was only included to run calculations, but ignored in the fitting procedure as its intensity is below the range of accuracy of our ML-IAP.
In all our classical spin-lattice dynamics calculations, our system size remained small compared to the typical magnetic domain-wall width in iron80. Thus, long-range dipole–dipole interactions could safely be neglected.
The parameters if this optimized spin Hamiltonian are contained in the Supplementary Table 1, along with LAMMPS input scripts used in the following section. The Supplementary Figure 3 also reports a comparison between our spin Hamiltonian and DFT data on the ΓN high-symmetry line, on which it was not parametrized. This additional calculation aims at probing the ability of our spin Hamiltonian to account for spin configurations that are outside of its training set.
SNAP potential
For this work, an interatomic potential for iron was developed that is specifically parameterized for use in coupled spin and molecular dynamics simulations. Training data for a Spectral Neighborhood Analysis Potential (SNAP)44,81,82 was collected to constrain the fit to the pressure and temperature phase space of <20 GPa and <2000 K. The set of non-colinear, spin-polarized VASP calculations includes α- (BCC), ϵ- (HCP) and liquid-iron, Table 1 displays the quantity of each training type and target properties that are captured therein. Optimization of a SNAP potential necessitates that the generated training database be broken into these groups (rows in Table 1) such that the weighted linear regression can (de-)emphasize different parts in search of a global minima in objective function errors. Each training group is assigned a unique weight for its’ associated energies and atomic forces for each candidate potential, optimization of these weights is controlled by DAKOTA. Regression is carried out using singular value decomposition with a squared loss function (L2 norm). In order to avoid double counting, and properly simulate the magnetic properties of iron in classical MD, we have adapted the SNAP fitting protocol44 to isolate the non-magnetic energy and forces from the generated training data. To do so, the fitted biquadratic spin Hamiltonian is used to evaluate the magnetic energy and forces for every atom in the training set, as well as the generated stress tensor pressure on the corresponding cell. Those quantities are then subtracted from the corresponding total DFT quantities. This is similar to previous uses of an ion core repulsion83 or electrostatic interaction term84 as a reference potential while fitting SNAP models. A key distinction, however, is that the inclusion of spin dynamics increase the dimensionality of the PES, which is not the case for Coulombic interactions. As a result, determination of magnetic energies and forces necessitates running a concurrent simulation for the spins within a standard MD run. The intricacies in evolving this 5N-dimensional system forward in time far exceed those encountered in the evaluation of static charge Coulombic interactions38.
The energy for a SNAP interatomic potential, \({E}_{{\mathrm{SNAP}}}^{i}\) for each atom i from its’ neighboring atom positions, rN, is expressed as a sum of the bispectrum components Bi
where the vector β are constant linear coefficients whose values are trained to reproduce energies and forces obtained from DFT training structures. Bispectrum components map the density correlations of neighboring atoms in a rotational and translation invariant manner, making them well posed descriptors of atomic energies and forces. By construction, the bispectrum components of an isolated atom are non-zero so the descriptors are shifted by the term B0 in order to force the potential energy to zero for an atom with no neighbors within the radial cutoff distance. Similarly, the forces on each atom k are expressed in terms of the derivative of atomic energies with respect to the position of atom k, where N is the total number of atoms in the structure
Optimization of the β terms in the SNAP potential was achieved using a single objective genetic algorithm within the DAKOTA software package43. Radial cutoff distance, training group weights and number of bispectrum descriptors were varied to minimize a set of objective functions, as percent error to available DFT or experimental85,86 data, that encapsulate the desired mechanical properties of Fe. These objective functions specific to α-iron are listed in Table 2, and the RMSE energy and force regression errors are included in optimization as well. In all objectives, our linear SNAP model with 30 bispectrum descriptors achieves accuracy in all mechanical properties within a few percent of experiment/DFT. Additionally, lattice constants and cohesive energies of γ- (FCC) and ϵ-iron (HCP) phases were fit, but given far less priority with respect to the α-iron mechanical properties resulting in ~6–7% errors with respect to DFT. Importantly, each of the objective functions were evaluated including the magnetic spin contributions to avoid unforeseen changes in property predictions. A full breakdown of the optimal training group weights and mean absolute energy/force errors are given in Table 1. Group weights listed have been adjusted by the number of configurations or forces they are applied to, therefore allowing for larger group weights to be (cautiously) interpreted more valuable at meeting the set of targeted objective functions. This optimized Fe-SNAP interatomic potential is detailed in the Supplementary Tables 1, 2, and 3 along with LAMMPS input scripts used in the following section.
Spin-lattice dynamics
Calculations are performed following the spin-lattice dynamics approach as implemented in the SPIN package of LAMMPS21,38, and set by the spin-lattice Hamiltonian below:
where \({{{{\mathcal{H}}}}}_{{\mathrm{mag}}}\) is the spin Hamiltonian defined by the combination of Eqs. (3) and (8). The term VSNAP(rij) is our SNAP ML-IAP. The second term on the right in Eq. (11), represents the kinetic energy, where the particle momentum is given as p and the mass of particle i is mi. Based on this spin-lattice Hamiltonian and leveraging the generalized spin-lattice Poisson bracket as defined by Yang et al.74, the equations of motion can be defined as:
Particle positions are advanced according to Eq. (12). The derivative of the momentum, given in Eq. (13), is dependent not only on the mechanical potential but the magnetic exchange functions as well. Here γL is the Langevin dam** constant for the lattice and f is a fluctuating force following Gaussian statistics given below38.
The fluctuating force f is coupled to γL via the fluctuation dissipation theorem as shown in Eq. (16). Here kB is the Boltzmann constant, Tl is the lattice temperature, and α and β are coordinates. Shown in Eq. (14) is the stochastic Landau-Lifshitz-Gilbert equation which describes the precessional motion of spins under the influence of thermal noise. In Eq. (14), λ is the transverse dam** constant and ωi is a spin force analog as shown in Eq. (5). Note that the gyromagnetic ratio is included in the calculation of the precession vectors (see Eq. (5)). The variable η(t) is a random vector whose components are drawn from a Gaussian probability distribution given below:
where the amplitude of the noise DS can be related to the temperature of the external spin bath Ts according to DS = 2πλkBTs/ℏ37.
SD-MD calculations are carried out using a 20 × 20 × 20 BCC cell. The BCC lattice is oriented along each of the coordinate directions. The MD timestep in all cases is set to 0.1 fs. The dam** constants are set to 0.1 (Gilbert dam**, no units) for the spin thermostat, and to 0.1 ps for the lattice thermostat. Initially all spins start out aligned in the z-direction. To measure the magnetic properties for the canonical ensemble we initially thermalize the system under NVT dynamics at the target spin/lattice temperatures for 40 ps and then sample the target properties for 10 ps using a sample interval of 0.001 ps. For pressure-controlled simulations (see PCC and MCPCC in the “Results” section), after the initial 40 ps of temperature equilibration we freeze the spin configuration and run isobaric-isothermal NPT dynamics in order to allow the system to thermally expand (still accounting for the effect of the magnetic pressure, generated by the spin Hamiltonian). The pressure dam** parameter is set to 10 ps. The pressure equilibration run is terminated once the system pressure drops below 0.05 GPa. After this, the spin configuration is unfrozen and another equilibration run is carried out under NVT dynamics for 20 ps. Unfreezing the spin configurations causes a small jump in the pressure, typically within the range of +/−2 GPa. To reduce this pressure fluctuation, a series of uniform isotropic box deformations are performed under the NVE ensemble. During this procedure the box is deformed in 0.02% increments every 2 ps until the magnitude of the pressure is reduced to negligible values (<10 MPa). Figure 6 displays the pressure profiles obtained within the FVC and PCMCC (similar to the PCC).

The FVC data is represented by the symbol/line combination  /
/ . Meanwhile, PCMCC data is represented by the symbol/line combination
. Meanwhile, PCMCC data is represented by the symbol/line combination  /
/ . Symbols, associated to the left axis, represent the pressure evolution in the FVC and PCMCC (similar to the PCC), as a function of lattice temperature. Doted lines, associated to the right axis, represent the spin thermostat temperature for the PCMCC and FVC (similar to PCC) as a function of the lattice temperature.
. Symbols, associated to the left axis, represent the pressure evolution in the FVC and PCMCC (similar to the PCC), as a function of lattice temperature. Doted lines, associated to the right axis, represent the spin thermostat temperature for the PCMCC and FVC (similar to PCC) as a function of the lattice temperature.
For the magnetization-controlled conditions (PCMCC in the “Results” section), the spin temperature is adjusted to match the experimental magnetization values. Spin temperature adjustments are made based on the magnetization curve obtained in the pressure-controlled conditions (PCC in the “Results” section). The corresponding spin-lattice temperature relationship is shown in Eqs. ((19)–(22)). Here the fitting coefficients are given as a1 = 471.6, a2 = 0.1, and α = 2.73, respectively. The functions Ts,pre and Ts,post prescribe how the spin temperature varies before and after the critical point. The form of Ts,pre is adopted from the spin temperature rescaling done by Evans et al.42. In our case we account for the shift in the Curie temperature when Ts = Tl by including the Tc,PCC = 576 K term in Eq. (20). The value of α found here agrees reasonably well with the work of Evans et al. where for iron α was found to be 2.876. At the critical point we use a switching function fsw to smoothly transition from Ts,pre to Ts,post. Above the experimental Curie temperature Tc,exp we do not assume Tl = Ts like Evans et al. The reason for this is that Tc,PCC ≠ Tc,exp hence if we assume Tl = Ts there will be a discontinuity in the effective temperature near the critical point which would also lead to unphysical discontinuities in the lattice constant and mechanical properties.
Figure 6 displays the spin temperature profiles for the FVC (and, similarly, the PCC), and the PCMCC. After the magnetic measurements we compute elastic constants by performing both uniaxial and shear deformations along each of the coordinate directions and planes. The magnitude of these deformations in all cases is 2% of the box length. Following each deformation the box is relaxed for 3 ps. After this relaxation the stresses are sampled for 2 ps using a sampling interval of 0.001 ps. For statistical averaging every spin-lattice dynamics simulation is ran six times using different random seeds. The final elastic and magnetic properties are averaged across these six simulations. The maximum uncertainty in all cases (FVC, PCC, and PCMCC) occurs near the critical points. For the elastic measurements and magnetization the maximum standard deviation is ~2% of the reported mean value. For the specific heat measurements the maximum standard of deviation occurs at the critical point and is ~25% of the reported mean value. Away from the critical point the standard of deviation for Cp is in the range of 5–10%.
In the Supplemental Note, we probe the ability of our magneto-elastic ML-IAP to be transferred to other material phases of iron. Supplementary Figures 1 and 2 display results obtained for liquid phase calculations as well as free energy measurements in the epsilon hcp phase of iron. Our result are in good agreement with DFT data and experimental observations. Those calculations constitute extrapolation of our potential (performed out of his training range), and are therefore not reported as main results of this manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
The code which was used to train the SNAP potential is available from https://github.com/FitSNAP/FitSNAP.
References
Tatsumoto, E. & Okamoto, T. Temperature dependence of the magnetostriction constants in iron and silicon iron. J. Phys. Soc. Jpn. 14, 1588–1594 (1959).
Bahl, C. R. H. & Nielsen, K. K. The effect of demagnetization on the magnetocaloric properties of gadolinium. J. Appl. Phys. 105, 013916 (2009).
Tavares, S., Fruchart, D., Miraglia, S. & Laborie, D. Magnetic properties of an AISI 420 martensitic stainless steel. J. Alloy. Compd. 312, 307–314 (2000).
Huang, S., Holmström, E., Eriksson, O. & Vitos, L. Map** the magnetic transition temperatures for medium-and high-entropy alloys. Intermetallics 95, 80–84 (2018).
Rao, Z. et al. Unveiling the mechanism of abnormal magnetic behavior of fenicomncu high-entropy alloys through a joint experimental-theoretical study. Phys. Rev. Mater. 4, 014402 (2020).
Jaime, M. et al. Piezomagnetism and magnetoelastic memory in uranium dioxide. Nat. Commun. 8, 1–7 (2017).
Nussle, T., Thibaudeau, P. & Nicolis, S. Dynamic magnetostriction for antiferromagnets. Phys. Rev. B 100, 214428 (2019).
Lejman, M. et al. Magnetoelastic and magnetoelectric couplings across the antiferromagnetic transition in multiferroic BiFeO3. Phys. Rev. B 99, 104103 (2019).
Patrick, C. E., Marchant, G. A. & Staunton, J. B. Spin orientation and magnetostriction of Tb1− xDyxFe2 from first principles. Phys. Rev. Appl. 14, 014091 (2020).
Graham, R., Morosin, B., Venturini, E. & Carr, M. Materials modification and synthesis under high pressure shock compression. Annu. Rev. Mater. Sci. 16, 315–341 (1986).
Surh, M. P., Benedict, L. X. & Sadigh, B. Magnetostructural transition kinetics in shocked iron. Phys. Rev. Lett. 117, 085701 (2016).
Moses, E. I., Boyd, R. N., Remington, B. A., Keane, C. J. & Al-Ayat, R. The national ignition facility: ushering in a new age for high energy density science. Phys. Plasmas 16, 041006 (2009).
Tschentscher, T. et al. Photon beam transport and scientific instruments at the European XFEL. Appl. Sci. 7, 592 (2017).
Tan, X., Chan, S., Han, K. & Xu, H. Combined effects of magnetic interaction and domain wall pinning on the coercivity in a bulk Nd60Fe30Al10 ferromagnet. Sci. Rep. 4, 6805 (2014).
Gràcia-Condal, A. et al. Multicaloric effects in metamagnetic Heusler Ni-Mn-In under uniaxial stress and magnetic field. Appl. Phys. Rev. 7, 041406 (2020).
Chandler, D. Introduction to Modern Statistical Mechanics (1987).
Horstemeyer, M. F. In Integrated Computational Materials Engineering (ICME) for Metals, Chap. 10, 410–423 (John Wiley & Sons, Ltd, 2012).
van der Giessen, E. et al. Roadmap on multiscale materials modeling. Model. Simul. Mater. Sci. Eng. 28, 043001 (2020).
Alder, B. J. & Wainwright, T. E. Studies in molecular dynamics. I. General method. J. Chem. Phys. 31, 459–466 (1959).
Rapaport, D. C. The Art of Molecular Dynamics Simulation (Cambridge University Press, 2004).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Computational Phys. 117, 1–19 (1995).
Voter, A. F., Montalenti, F. & Germann, T. C. Extending the time scale in atomistic simulation of materials. Annu. Rev. Mater. Res. 32, 321–346 (2002).
Zepeda-Ruiz, L. A., Stukowski, A., Oppelstrup, T. & Bulatov, V. V. Probing the limits of metal plasticity with molecular dynamics simulations. Nature 550, 492–495 (2017).
Huan, T. D. et al. A universal strategy for the creation of machine learning-based atomistic force fields. npj Comput. Mater. 3, 1–8 (2017).
Smith, J. S., Isayev, O. & Roitberg, A. E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 8, 3192–3203 (2017).
Zhang, L., Han, J., Wang, H., Car, R. & E, W. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Jaramillo-Botero, A., Naserifar, S. & Goddard, W. A. General multiobjective force field optimization framework, with application to reactive force fields for silicon carbide. J. Chem. Theory Comput. 10, 1426–1439 (2014).
Lubbers, N., Smith, J. S. & Barros, K. Hierarchical modeling of molecular energies using a deep neural network. J. Chem. Phys. 148, 241715 (2018).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Computational Phys. 285, 316–330 (2015).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Li, X.-G., Chen, C., Zheng, H., Zuo, Y. & Ong, S. P. Complex strengthening mechanisms in the Mo-V-Nb-Ti-Zr multi-principal element alloy. npj Computat. Mater. 6, 1–10 (2020).
Cusentino, M., Wood, M. & Thompson, A. Suppression of helium bubble nucleation in beryllium exposed tungsten surfaces. Nucl. Fusion 60, 126018 (2020).
Dragoni, D., Daff, T. D., Csányi, G. & Marzari, N. Achieving DFT accuracy with a machine-learning interatomic potential: thermomechanics and defects in bcc ferromagnetic iron. Phys. Rev. Mater. 2, 013808 (2018).
Ma, P.-W., Woo, C. & Dudarev, S. Large-scale simulation of the spin-lattice dynamics in ferromagnetic iron. Phys. Rev. B 78, 024434 (2008).
Ma, P.-W., Dudarev, S. & Woo, C. SPILADY: a parallel CPU and GPU code for spin–lattice magnetic molecular dynamics simulations. Computer Phys. Commun. 207, 350–361 (2016).
Ma, P.-W. & Dudarev, S. Atomistic spin-lattice dynamics. Handbook of Materials Modeling: Methods: Theory and Modeling 1017–1035 (2020).
Tranchida, J., Plimpton, S., Thibaudeau, P. & Thompson, A. P. Massively parallel symplectic algorithm for coupled magnetic spin dynamics and molecular dynamics. J. Computat. Phys. 372, 406–425 (2018).
Dos Santos, G. et al. Size- and temperature-dependent magnetization of iron nanoclusters. Phys. Rev. B 102, 184426 (2020).
Zhou, Y., Tranchida, J., Ge, Y., Murthy, J. & Fisher, T. S. Atomistic simulation of phonon and magnon thermal transport across the ferromagnetic-paramagnetic transition. Phys. Rev. B 101, 224303 (2020).
Ma, P.-W., Dudarev, S. & Wróbel, J. S. Dynamic simulation of structural phase transitions in magnetic iron. Phys. Rev. B 96, 094418 (2017).
Evans, R. F., Atxitia, U. & Chantrell, R. W. Quantitative simulation of temperature-dependent magnetization dynamics and equilibrium properties of elemental ferromagnets. Phys. Rev. B 91, 144425 (2015).
Eldred, M. S. et al. DAKOTA, a multilevel parallel object-oriented framework for design optimization, parameter estimation, uncertainty quantification, and sensitivity analysis: version 5.0 user’s manual. Sandia National Laboratories, Tech. Rep., Citeseer SAND2010-2183 (2006).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015).
Ashcroft, N. W., Mermin, N. D. & Wei, D. Solid State Physics (Cengage Learning Asia PTE Limited, 2016).
Dragoni, D., Ceresoli, D. & Marzari, N. Thermoelastic properties of α-iron from first-principles. Phys. Rev. B 91, 104105 (2015).
Dragoni, D., Ceresoli, D. & Marzari, N. Vibrational and thermoelastic properties of bcc iron from selected eam potentials. Comput. Mater. Sci. 152, 99–106 (2018).
Körmann, F. et al. Free energy of bcc iron: Integrated ab initio derivation of vibrational, electronic, and magnetic contributions. Phys. Rev. B 78, 033102 (2008).
Arakawa, K. et al. Quantum de-trap** and transport of heavy defects in tungsten. Nat. Mater. 19, 508–511 (2020).
Rosengaard, N. & Johansson, B. Finite-temperature study of itinerant ferromagnetism in Fe, Co, and Ni. Phys. Rev. B 55, 14975 (1997).
Zimmermann, B. et al. Comparison of first-principles methods to extract magnetic parameters in ultrathin films: Co/Pt (111). Phys. Rev. B 99, 214426 (2019).
Drautz, R. & Fähnle, M. Spin-cluster expansion: parametrization of the general adiabatic magnetic energy surface with ab initio accuracy. Phys. Rev. B 69, 104404 (2004).
Drautz, R. Atomic cluster expansion of scalar, vectorial, and tensorial properties including magnetism and charge transfer. Phys. Rev. B 102, 024104 (2020).
Marinica, M.-C., Willaime, F. & Crocombette, J.-P. Irradiation-induced formation of nanocrystallites with C15 laves phase structure in bcc iron. Phys. Rev. Lett. 108, 025501 (2012).
Chapman, J. B., Ma, P.-W. & Dudarev, S. L. Effect of non-Heisenberg magnetic interactions on defects in ferromagnetic iron. Phys. Rev. B 102, 224106 (2020).
Chapman, J. B., Ma, P.-W. & Dudarev, S. L. Dynamics of magnetism in Fe-Cr alloys with Cr clustering. Phys. Rev. B 99, 184413 (2019).
Klaver, T., Drautz, R. & Finnis, M. Magnetism and thermodynamics of defect-free Fe-Cr alloys. Phys. Rev. B 74, 094435 (2006).
Kalantar, D. et al. Direct observation of the α-ε transition in shock-compressed iron via nanosecond X-ray diffraction. Phys. Rev. Lett. 95, 075502 (2005).
Woo, C., Wen, H., Semenov, A., Dudarev, S. & Ma, P.-W. Quantum heat bath for spin-lattice dynamics. Phys. Rev. B 91, 104306 (2015).
Bergqvist, L. & Bergman, A. Realistic finite temperature simulations of magnetic systems using quantum statistics. Phys. Rev. Mater. 2, 013802 (2018).
Novikov, I., Grabowski, B., Kormann, F. & Shapeev, A. Machine-learning interatomic potentials reproduce vibrational and magnetic degrees of freedom. Preprint at https://arxiv.org/abs/2012.12763 (2020).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Halilov, S., Perlov, A., Oppeneer, P. & Eschrig, H. Magnon spectrum and related finite-temperature magnetic properties: a first-principle approach. EPL 39, 91 (1997).
Kurz, P., Förster, F., Nordström, L., Bihlmayer, G. & Blügel, S. Ab initio treatment of noncollinear magnets with the full-potential linearized augmented plane wave method. Phys. Rev. B 69, 024415 (2004).
Sandratskii, L. Noncollinear magnetism in itinerant-electron systems: theory and applications. Adv. Phys. 47, 91–160 (1998).
Marsman, M. & Hafner, J. Broken symmetries in the crystalline and magnetic structures of γ-iron. Phys. Rev. B 66, 224409 (2002).
Szilva, A. et al. Interatomic exchange interactions for finite-temperature magnetism and nonequilibrium spin dynamics. Phys. Rev. Lett. 111, 127204 (2013).
Kaneyoshi, T. Introduction to Amorphous Magnets (World Scientific Publishing Company, 1992).
Yosida, K., Mattis, D. C. & Yosida, K. THEORY OF MAGNETISM.: Edition en anglais, Vol. 122 (Springer Science & Business Media, 1996).
Pajda, M., Kudrnovskỳ, J., Turek, I., Drchal, V. & Bruno, P. Ab initio calculations of exchange interactions, spin-wave stiffness constants, and Curie temperatures of Fe, Co, and Ni. Phys. Rev. B 64, 174402 (2001).
Yang, K.-H. & Hirschfelder, J. O. Generalizations of classical poisson brackets to include spin. Phys. Rev. A 22, 1814 (1980).
Thompson, A. P., Plimpton, S. J. & Mattson, W. General formulation of pressure and stress tensor for arbitrary many-body interaction potentials under periodic boundary conditions. J. Chem. Phys. 131, 154107 (2009).
Evans, R. F. Handbook of Materials Modeling: Applications: Current and Emerging Materials 427–448 (2020).
Leger, J., Loriers-Susse, C. & Vodar, B. Pressure effect on the Curie temperatures of transition metals and alloys. Phys. Rev. B 6, 4250 (1972).
Morán, S., Ederer, C. & Fähnle, M. Ab initio electron theory for magnetism in Fe: pressure dependence of spin-wave energies, exchange parameters, and Curie temperature. Phys. Rev. B 67, 012407 (2003).
Körmann, F., Dick, A., Hickel, T. & Neugebauer, J. Pressure dependence of the Curie temperature in bcc iron studied by ab initio simulations. Phys. Rev. B 79, 184406 (2009).
Skomski, R. et al. Simple Models of Magnetism (Oxford University Press on Demand, 2008).
Wood, M. A. & Thompson, A. P. Extending the accuracy of the snap interatomic potential form. J. Chem. Phys. 148, 241721 (2018).
Zuo, Y. et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 124, 731–745 (2020).
Wood, M. A., Cusentino, M. A., Wirth, B. D. & Thompson, A. P. Data-driven material models for atomistic simulation. Phys. Rev. B 99, 184305 (2019).
Deng, Z., Chen, C., Li, X.-G. & Ong, S. P. An electrostatic spectral neighbor analysis potential for lithium nitride. npj Comput. Mater. 5, 1–8 (2019).
Goryaeva, A. M., Maillet, J.-B. & Marinica, M.-C. Towards better efficiency of interatomic linear machine learning potentials. Computat. Mater. Sci. 166, 200–209 (2019).
Adams, J. J., Agosta, D., Leisure, R. & Ledbetter, H. Elastic constants of monocrystal iron from 3 to 500 k. J. Appl. Phys. 100, 113530 (2006).
Wallace, D. C., Sidles, P. & Danielson, G. Specific heat of high purity iron by a pulse heating method. J. Appl. Phys. 31, 168–176 (1960).
Touloukian, Y. & Buyco, E. Thermophysical Properties of Matter, Vol. 4, specific heat (IFI/Plenum, 1970).
Crangle, J. & Goodman, G. The magnetization of pure iron and nickel. Proc. R. Soc. Lond. A. Math. Phys. Sci. 321, 477–491 (1971).
Seki, I. & Nagata, K. Lattice constant of iron and austenite including its supersaturation phase of carbon. ISIJ Int. 45, 1789–1794 (2005).
Basinski, Z. S., Hume-Rothery, W. & Sutton, A. The lattice expansion of iron. Proc. R. Soc. Lond. Ser. A. Math. Phys. Sci. 229, 459–467 (1955).
Loong, C.-K., Carpenter, J., Lynn, J., Robinson, R. & Mook, H. Neutron scattering study of the magnetic excitations in ferromagnetic iron at high energy transfers. J. Appl. Phys. 55, 1895–1897 (1984).
Lynn, J. Temperature dependence of the magnetic excitations in iron. Phys. Rev. B 11, 2624 (1975).
Acknowledgements
All authors thank Mark Wilson for his detailed review and edits. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. A.C. acknowledges funding from the Center for Advanced Systems Understanding (CASUS) which is financed by the German Federal Ministry of Education and Research (BMBF) and by the Saxon State Ministry for Science, Art, and Tourism (SMWK) with tax funds on the basis of the budget approved by the Saxon State Parliament.
Author information
Authors and Affiliations
Contributions
A.C., M.A.W., M.P.D., and J.T. performed the DFT calculations. J.B.M., M.C.M., J.T., and M.A.W. generated the Database of configurations. J.T. implemented the extended spin Hamiltonian and the magnetic pressure computation in LAMMPS, and parametrized it on first-principles calculations. M.A.W. and S.N. trained the SNAP potential. J.T. and S.N. performed the magneto-static calculations. S.N., A.P.T., M.A.W., and J.T. performed the magneto-dynamics calculations. All authors participated in conceiving the research and writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nikolov, S., Wood, M.A., Cangi, A. et al. Data-driven magneto-elastic predictions with scalable classical spin-lattice dynamics. npj Comput Mater 7, 153 (2021). https://doi.org/10.1038/s41524-021-00617-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-021-00617-2
- Springer Nature Limited
This article is cited by
-
Non-collinear magnetic atomic cluster expansion for iron
npj Computational Materials (2024)
-
Spin-lattice-dynamics analysis of magnetic properties of iron under compression
Scientific Reports (2023)
-
Constrained DFT-based magnetic machine-learning potentials for magnetic alloys: a case study of Fe–Al
Scientific Reports (2023)
-
A self-adaptive first-principles approach for magnetic excited states
Quantum Frontiers (2023)
-
Magnetic Moment Tensor Potentials for collinear spin-polarized materials reproduce different magnetic states of bcc Fe
npj Computational Materials (2022)