

Abstract
Postzygotic reproductive isolation, which results in the irreversible divergence of species, is commonly accompanied by hybrid sterility, necrosis/weakness, or lethality in the F1 or other offspring generations. Here we show that the loss of function of HWS1 and HWS2, a couple of duplicated paralogs, together confer complete interspecific incompatibility between Asian and African rice. Both of these non-Mendelian determinants encode the putative Esa1-associated factor 6 (EAF6) protein, which functions as a characteristic subunit of the histone H4 acetyltransferase complex regulating transcriptional activation via genome-wide histone modification. The proliferating tapetum and inappropriate polar nuclei arrangement cause defective pollen and seeds in F2 hybrid offspring due to the recombinant HWS1/2-mediated misregulation of vitamin (biotin and thiamine) metabolism and lipid synthesis. Evolutionary analysis of HWS1/2 suggests that this gene pair has undergone incomplete lineage sorting (ILS) and multiple gene duplication events during speciation. Our findings have not only uncovered a pair of speciation genes that control hybrid breakdown but also illustrate a passive mechanism that could be scaled up and used in the guidance and optimization of hybrid breeding applications for distant hybridization.
Similar content being viewed by others

Introduction
Reproductive isolation (RI) hinders gene flow between intraspecific or interspecific populations during speciation, therefore contributing to the maintenance of species identity1,2. Hybrid breakdown (HB), a typical form of RI, is defined as reduced hybrid viability and/or fertility segregation in the F2 or the later generation. Differing from other hybrid incompatibilities, HB involves intrinsic postzygotic reproductive barriers, such as hybrid inviability (including weakness3, necrosis4, and chlorosis5) and hybrid sterility (in the male, female, or both gametes). The classical Bateson-Dobzhansky-Muller (BDM) model partially addresses hybrid sterility, the most common form of postzygotic barrier that is attributed to deleterious genetic interactions between incompatible allelic variations, but fails to consider the divergent evolution process6,7,8. A widely accepted viewpoint that has been proposed to reasonably explain the biased allele transmission and disharmonious effect conferring plant hybrid sterility is the killer-protector system9,10. In broad terms, genomic conflicts involved in hybrid incompatibility that occurs in sporophytes and gametophytes are basically divided into two types; single or dual locus-controlled F1/F2 male/female sterility models11,Full size image
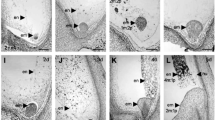
Ultrastructural studies of the tapetal layer showed that the tapetal membrane of WYJ7 was distinctly decomposed and precipitated around Ubisch bodies at stage 10 (Fig. 2m). A typical two-layer wall structure composed of the tectum, bactum, and nexine was also clearly visible (Figs. 2n, n1). In contrast, in the chamber of sterile anthers of NIL-hws1WYJ7/hws2CG14 plants, there were no clear and intact Ubisch bodies observed on the inner tangential wall of the tapetum (Fig. 2o). Remarkably large vacuoles in the thickened tapetum were irregularly formed during the process of tapetal disintegration (Fig. 2o). Moreover, parts of the exine in microspores showed abnormal protuberances with uneven bactum (Figs. 2p, p1). Consistent with the above observations, the main cytological reason behind pollen abortion is considered to be the stimulated proliferation of the tapetal cells, which interferes with microspore development. Scanning electron microscopy (SEM) observations further visualized the overall appearance features of anthers and pollen grains at maturity, revealing that abortion in hws1WY7J/hws2CG14-type pollen grains is possibly due to their shrunken morphology and poorly-developed anther wall (Figs. 2q, q1-q8).
Dynamic examination of embryo sac development in NIL-hws1WYJ7/hws2CG14 plants showed no obvious defects with respect to structure and morphology prior to the eight-nucleate embryo sac stage (Fig. 2r, s). However, the two polar nuclei occasionally failed to correctly locate and form a binucleate central cell near the micropylar end during later stages of development (Fig. 2t–w). These abortive and smaller embryo sacs were also found to be developmentally arrested, either empty or degenerated, in comparison with those in the fertile control (WYJ7) plants (Fig. 2v, w). Based on these results, we concluded that inappropriate polar nuclei arrangement, stagnant development, and abnormal cellularization could result in the inability to form mature embryo sacs during female gamete development in NIL-hws1WYJ7/hws2CG14 plants.
Dysfunctions in HWS1/2 contribute to H4Ac loss and transcriptomic reprogramming misregulation
EAF6 is a critical component of the nucleosomal acetyltransferase of the H3/H4 (NuA3/NuA4) complex and has been reported to participate in extensive transcriptional regulation via nuclear H4 or H2A acetylation, thereby affecting plant viability38,39,40. Subcellular localization analysis indicated that HWS1 and HWS2 are specifically targeted to the nuclei (Fig. 3a). The in vitro histone acetylation (HAT) assay showed that HWS2 is able to acetylate a histone H4 peptide (Fig. 3b). Fractionation of chromatin followed by western blotting using antibodies against various acetylation sites in the histone H2A, H3 and H4 N-terminal tail showed a pronounced decline in acetyl-H2A and acetyl-H4 in NIL-hws1WYJ7/hws2CG14, and this decline was most prominent at lysine 5 and 9 of histone H2A (H2AK5Ac and H2AK9Ac) and at lysine 5 of histone H4 (H4K5Ac) among all eight histone H2A and H4 lysine residues tested (Fig. 3c). To gain more insight into the correlation between the loss of H4Ac and transcriptional reprogramming, we first performed RNA deep sequencing (RNA-seq) and identified 19,806 differentially-expressed genes (DEGs) (fold-change >2) between the two corresponding NILs (Supplementary Fig. 7a). The enriched terms from combined Gene Ontology (GO) classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis showed that the statistically significant DEGs were primarily associated with mRNA surveillance, fatty acid, lipid metabolism, RNA transport, and small molecule binding process (Supplementary Fig. 7b, c). As shown by heat maps, the expression levels of several representative male determinants related to fatty acid synthesis and lipid transport were substantially down-regulated in NIL-hws1WYJ7/hws2CG14 plants (Supplementary Fig. 7d)41,42,43,44,45, which may account for the adverse formation of the enlarged tapetal zone, unshaped pollen exine, and unusual cuticle surface.

a Subcellular localization of HWS1 and HWS2 fused with the yellow fluorescent protein (YFP) reporter. A nuclear localization signal marker (NLS-DsRed) was co-expressed with the HWS1/2-YFP fusion proteins in rice protoplasts. Scale bars = 10 µm. b HAT activity testing of HWS2 towards histone H4 by fluorometry. AFU, Arbitrary Fluorescence Units. c Endogenous acetylated histone H2A, H3, and H4 levels and substrate specificity determined by western blotting. The specific tested antibodies are shown on the left and the differences in visible band intensity are indicated by red arrowheads. Lane #1: NIL-hws1WYJ7/hws2CG14; Lane #2: NIL-HWS1CG14/hws2CG14; Lane #3: NIL-hws1WYJ7/HWS2WYJ7; and Lane #4: NIL-HWS1CG14/HWS2WYJ7. d The distribution profile of averaged H4Ac occupancy across the gene body in NIL-HWS1CG14/hws2CG14 and NIL-hws1WYJ7/hws2CG14 (****p < 0.0001, calculated by two-sided Welch’s t-test). e Venn diagram showing the overlap between RNA-seq differentially-regulated genes with reduced expression levels and ChIP-seq differentially-enriched genes with reduced H4Ac levels. f KEGG enrichment bubble plot of the overlap** DEG set in e. All significantly enriched (FDR < 0.05) KEGG terms are shown while the emphasized pathways are additionally indicated with a green background. g Genome browser views of mRNA and H4Ac signals of gene examples selected for validation. h qRT-RCR expression analysis of two NADH-dependent enoyl-ACP reductase (LOC_Os08g23810 and LOC_Os09g10600) and OsKASI (LOC_Os06g09630) in the young panicles of NIL-HWS1CG14/hws2CG14 and NIL-hws1WYJ7/hws2CG14 plants. The expression level of each tested gene normalized to rice Ubiquitin in NIL-HWS1CG14/hws2CG14 was set to 1.0. i ChIP-qPCR analysis showing the relative enrichment of indicated genes identified in h. j, k, Measurement of biotin level in NIL-hws1WYJ7/hws2CG14 versus NIL-HWS1CG14/hws2CG14 plants (j) and NIL-hws1WYJ7/hws2CG14 versus NIL-hws1WYJ7/HWS2WYJ7 plants (k). Data in b, h, i, j, and k are mean ± SEM (n = 3 (b, h, and i) or 4 (j and k) biological replicates). In b, different letters denote significant differences (p < 0.05, one-way ANOVA with two-sided Tukey’s HSD test), p values are adjusted and shown in the Source Data file. A two-sided Student’s paired t-test was used to generate the p-values in h–k. The experiments in a and c were repeated three times independently, with similar results.
To further visualize the in vivo differences in the degree of acetylated histone H4 at the whole-genome level, a chromatin immunoprecipitation sequencing (ChIP-seq) experiment was unitedly employed to analyze the transcriptome data. Strikingly, the average H4 occupancy in NIL-hws1WYJ7/hws2CG14 displayed a dramatic suppression within the peak downstream of the transcriptional start site (TSS) (Fig. 3d and Supplementary Fig. 7e). A total of 2,297 H4Ac enriched peaks corresponding to 2,175 genes were identified, the vast majority of which were down-regulated and localized at the promoter (88.98%) (Supplementary Fig. 7f). The principal binding site detected in the enriched peaks was the ORB2/RRM-type motif (GGATTTGGGG) (Supplementary Fig. 7g), which basically recruits RNA-binding proteins involved in post-transcriptional regulation.
We hereupon defined the dysregulated genes with H4Ac enrichment peaks at the promoter and/or gene body regions as candidate HWS1/2 target genes (Fig. 3e). Among the categories that overlap with those of the transcriptome datasets, genes related to vitamin metabolism (biotin and thiamine), fatty acid biosynthesis, ether lipid metabolism, and carbohydrate metabolism with reduced H4Ac levels showed altered expression (Fig. 3f and Supplementary Fig. 7h). We, therefore, selected some well-characterized sterility genes belonging to these particular pathways with a strong and uniform reduction in sequencing windows to examine their mRNA and H4Ac levels as validation (Fig. 3g)46. We examined the expression of these potential genes by qRT-PCR assays, and all of them showed greatly decreased expression in NIL-hws1WYJ7/hws2CG14 plants, which is in line with the results of the transcriptional profile (Fig. 3h). In parallel, chromatin immunoprecipitation quantitative polymerase chain reaction (ChIP-qPCR) results also showed a relatively lower level of histone H4 acetylation in these certain genomic regions (Fig. 3i), implying that in the absence of HWS1/2, the chromatin status is not appropriately established. Besides, determined quantities of biotin were significantly decreased in NIL-hws1WYJ7/hws2CG14 plants in contrast with NIL-HWS1CG14/hws2CG14 (Fig. 3j) and NIL-hws1WYJ7/HWS2WYJ7 (Fig. 3k) plants. Taken together, our findings suggest that HWS1/2 is indispensable for NuA4-dependent H4 acetylation and gene transcriptional activation by proper epigenetic modifications, and is especially essential for gametophyte development through the maintenance of fatty acid, lipid, and vitamin homeostasis.
Evolutionary trajectory and EAF6 gene duplication events in Oryza species
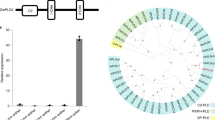
To trace the evolutionary origins of HWS1 and HWS2 (OsEAF6), we performed a collinearity analysis around these two distinct loci among some representative Oryza species and other distantly related gramineous crop species (Fig. 4a and Supplementary Fig. 8a–d). Comparative genomics showed that there is widespread synteny in the grass family, but that EAF6 is a single-copy gene in O.punctata (BB genome), O.officinalis (CC genome), O.brachyantha (FF genome) (Fig. 4a), and the majority of AA genome wild progenitors (Supplementary Fig. 8a, b), indicating that the duplication of EAF6 pairs may have evolved de novo in the O.sativa and was distinguished from the other outgroup species Zea mays (maize), Hordeum vulgare (barley), and Zizania latifolia (Manchurian wild rice) (Fig. 4a). These sister groups to Oryza mostly showed a common block of homologous synteny around the EAF6 region retained on chromosome 2 and shared a compatible lineage with their closest wild rice relative (Leersia perrieri) aside from the non-overlap** lineage sorting in AA-genome Oryza species (Fig. 4a and Supplementary Fig. 8a, b). This suggests that the two copies of OsEAF6 in O.sativa likely originated from their ancestral EAF6 gene located in a given position of chromosome 2 through speciation, genetic duplication, or horizontal gene transfer. In addition, we made further local collinearity analysis of HWS1 between O.sativa and the other five grasses (Supplementary Fig. 8c) and HWS2 (Supplementary Fig. 8d) between O.sativa, O.officinalis, and L.perrieri. The results showed that the chromosome segment exhibited significant syntenic relationships from Zea may to Oryza sativa, but it seemed that HWS1 came out of nowhere (Supplementary Fig. 8c). On the contrary, the collinearity of the HWS2 region in related species is much worse, indicating a possibly huge chromosome structure variation in HWS2 locus (Supplementary Fig. 8d). Given that transposable elements (TEs) generally cause no synteny32, we speculated that HWS1/2 gene pair may be generated by transposon-mediated gene replication. Considering the abundant transposon insertions located nearby, we carried out genome-wide transposon annotation analysis and showed that the segmental genomic duplication carrying OsEAF6 was probably generated by DNA transposon-mediated gene replication (Supplementary Fig. 9)47. To further clarify the evolution process of HWS1/2, we constructed the phylogenetic tree and found that the gene tree of the HWS1 locus was incongruent with the species tree48 in the certain HWS1 allele (hws1C) (Fig. 4b). We next generated the gene tree of HWS1 by using the 133 species (Supplementary Fig. 10a and Supplementary Data 1), and the result showed that all the African rice were more closely to nearly all the japonica and some of the O.rufipogon rice (Supplementary Fig. 10a).

a Comparative genomes and syntenic gene analysis of HWS1/2 in rice and selected Gramineae species. HWS1/2 orthologs are marked with red dots on the chromosomes in each species. Orange lines indicate conserved syntenic blocks harboring EAF6 (HWS1/2). b A phylogenetic tree showing the relationships among diploid species of Oryza based on EAF6 sequence variation. The EAF6 copies from each species are shown with colored backgrounds. Asterisks and green lines represent branch of the gene tree that is inconsistent with the species tree. c Diagram shows the distinguishment between ILS and introgression scenarios by using the estimated divergence times. d, e The species trees (upper panel) and gene trees (low panel) with divergence time to distinguish between ILS and introgression. Representative varieties of Or-I and Or-III with these two topological structures were selected to estimate the species differentiation time and the differentiation time of HWS1 gene. The green and blue lines indicate the ILS events. The estimated divergence times of speciation (t) and ILS (tIL) are colored in green in Or-III_O.barthii topology and blue in Or-I_O.barthii topology. f Ks distribution of HWS1/2 in Classes I and Classes II of the 133 selected Asian and African accessions or cultivars relative to the HWS1 gene on chromosome 1 of O.meridionalis. The taxonomic groups are demarcated by dashed lines. g Expression analysis of HWS1 and HWS2 among the representative rice varieties in the three classes (n = 6, 15, and 9 in Classes I, II, and III, respectively) based on RNA-seq data. The HWS1 and HWS2 alleles are indicated by red and black bars, respectively. TPM, Transcripts Per kilobase per Million mapped reads. All the TPM values are listed in Supplementary Data 2. h A sliding window analysis of π values across a 200 kb chromosomal region including sequences upstream and downstream of HWS1 in japonica and indica varieties and three types of wild rice (Or) using published data. The position of the HWS1 locus is framed with a dotted box and marked with a red arrowhead.
Although these phylogenetic discrepancies potentially stem from introgression or ILS, coalescence times for regions under ILS should be earlier than the speciation event while introgression occurs later32(Fig. 4c). To distinguish which event leads to the incongruence of gene tree and species tree, we first compared the species divergence time (t) with expected coalescence time under ILS (tIL) which was estimated based on the gene tree of HWS1. Since O.rufipogon could be divided into three groups: Or-I/II/III and Or-I is the ancestor of indica while Or-III is the ancestor of japonica49, we therefore selected the representative varieties and divided into two major paired-topology categories, W1970 (HWS1T)_W1536 (hws1C)_NH280_W1952 (Fig. 4d) and W1547 (hws1C)_W3012 (HWS1T)_NH280_W1952 (Fig. 4e), both of which belong to Or-I, Or-III, O.barthii, and O.meridionalis, respectively. The hws1C gene of W1536 (Or-III) and W1547 (Or-I) was more closely related to HWS1T of O.barthii and did not match the species tree (Fig. 4d, e). Next, we calculated the synonymous substitution rate (Ks) of 3071 single-copy gene pairs between W1970 (Or-I) and W1536 (Or-III), as well as between W1547 (Or-I) and W3012 (Or-III), and estimated the differentiation time of the two species to be 0.062 and 0.056 MYA, respectively (Fig. 4d, e and Supplementary Fig. 10b, c). Likewise, we calculated the Ks values of the HWS1 gene pairs between Or-III (hws1C) and O.barthii (HWS1T), along with Or-I (hws1C) and O.barthii (HWS1T), both of their estimated differentiation time was 0.92 MYA (Fig. 4d, e). The coalescence time of the HWS1 gene was significantly earlier than the corresponding speciation time (Fig. 4d, e). This strongly suggests that ILS, not introgression, may be probably the major reason for the pervasive signatures of incongruence in these certain HWS1 alleles (hws1C). To further exclude the introgression event, we conducted the four-taxon D-statistic test with the ABBA-BABA method. No obvious introgression signal was observed in each of the three paired-topology categories ((Or-III, Or-I)_O.barthii_O.meridionalis, (Or-II, Or-III)_O.barthii_O.meridionalis, and (Or-II, Or-I)_O.barthii_O.meridionalis) in the window where HWS1 gene was located (Supplementary Fig. 10d).
To comprehensively investigate the evolution and duplication processes of the HWS1/2 alleles, we selectively compared the genomic sequences among a diverse group of AA-genome rice species. This analysis included wild and domesticated Asian and African rice but excluded a group of certain rice cultivars/accessions due to the presence of multiple tandemly-repeated HWS2 loci (Supplementary Fig. 9b and Supplementary Data 1). Based on the pan-genome data, we calculated the synonymous substitution (Ks) values of HWS1/2 from 133 selected varieties relative to the HWS1 of O.meridionalis. This analysis identified three evolution processes (Ks = 0.028, 0.035, and 0.053, respectively) of the most HWS1/2 alleles (Fig. 4f). The HWS1 of O.rufipogon and O.barthii were diverged from O.meridionalis (Ks = 0.028 and 0.053), and then the HWS2 (HWS2T) gene in O.rufipogon was duplicated from HWS1 (HWS1T) (Ks = 0.035) (Fig. 4f), while some of the HWS1 allele (hws1C) in O.rufipogon varieties experienced the ILS event (Ks = 0.049) (Fig. 4c–e and Supplementary Fig. 10b–e). Notably, the transcriptional expression profile further revealed that the HWS1 genes all underwent gene inactivation resulting from expression failure in certain O.rufipogon and japonica (hws1C) (Fig. 4g and Supplementary Data 2). In the three wild rice populations, nucleotide diversity (π) in adjacent regions of HWS1 gene (7.35 Mb–7.4 Mb) was significantly higher than that of cultivated rice, indicating strong positive selection in this region (Fig. 4h). Evidently, a relatively low π value for the entire 200-kb region was also detected in the japonica population (Fig. 4h), indicating the presence of a bottleneck effect shaped by demographic and selective pressure. Nevertheless, the expression failure of HWS1 in nearly all japonica might be due to the founder effect resulting from the domestication of japonica rice. Taken together, HWS1/2, the functionally redundant paralogs, are thought to be duplicated and originated from a common ancestor in each divergent lineage, and the shared genetic variation between the certain O.rufipogon and O.barthii may be likely attributed to ILS.
Variation analysis and functional classification of the HWS1/2 genes in rice
Using the nucleotide variation identified in the HWS1/2 gene bodies and the 3’ and 5’ untranslated regions (UTRs), we performed an association test with a local genomic scan and uncovered several mutations that are correlated with gene expression in the same linkage disequilibrium (LD) block (Supplementary Data 2 and Supplementary Data 3). The strongest signal was present at the SNP1_7385301 site, a T-to-C substitution located in the upstream region that is inferred to be a proposed causative variant in HWS1 (Fig. 5a and Supplementary Fig. 4c). To verify whether the T/C variant in the HWS1 promoter affects the gene expression, we generated a range of promoter mutation constructs, each fused with LUC (luciferase) reporter gene and introduced into rice protoplasts to monitor promoter activity (Fig. 5b). The quantitation of firefly luciferase expression showed that the HWS1CG14pro: Luc activity was dramatically reduced after the point mutation (“T” to “C”) in SNP1_7385301 site. Instead, the “C” to “T” replacement in hws1WYJ7 rescued the expression failure of HWS1 in WYJ7, indicating that this core mutation may exert a great impact on the level of promoter activity. Several Asian cultivars including four japonica and three indica varieties were crossed with SG178 to determine the responsible polymorphic site (Fig. 5c). Plants exhibiting sterility and weakness were observed in the F2 populations derived from crosses between C-carrying varieties and SG178 (Fig. 5d–g). On the contrary, when carrying the T allele of SNP1_7385301, the functional single nucleotide polymorphism (SNP), all of the descendants with different genotypes showed full seed set (Fig. 5h–j). In summary, our experiments in which we crossed WYJ7 and SG178 with a wide range of rice varieties showed that the targeted variation in HWS1 accounts for the interspecific sterility in rice. This potentially functional allele is referred to as HWS1T while the sterile allele lacking promoter activity or the syntenic segmental block is called hws1C or hws2Null, both of which represent a loss-of-function type (Fig. 4g and Supplementary Data 2). Further sequence comparisons revealed that the rice accessions/varieties could be divided into three major HWS groups; Class I (HWS1T/HWS2), Class II (HWS1T/hws2Null), and Class III (hws1C/HWS2), and all three groups contained O. rufipogon accessions (Fig. 5k).

a Case/control test for SNPs in the 4.6 kb fine-mapped region around the HWS1 locus and expression level using a standard one-degree-of-freedom allelic Chi-square test in Plink. Local Manhattan plot (top), gene structure (middle), and LD heatmap (bottom) surrounding HWS1 are shown. The red diamond in the Manhattan plot shows the position of the proposed causative mutation SNP (SNP1_7385301). P values and identified SNPs are presented in Supplementary Data 3. b Activity analysis of HWS1 promoter. Upper part: schematic diagram of the pGREENII 0800-LUC vector fused with HWS1 promoter. Leftward part: a series of constructs with site-directed mutation HWS1 promoter. S1, 3000-bp promoter region of HWS1CG14, S2, T to C point mutation of S1; S3, 3000-bp promoter region of hws1WYJ7; S4, C to T point mutation of S3. Rightward part: expression levels of firefly luciferase normalized by Renilla luciferase. c Haplotypes of HWS1 of various germplasm. The identified SNPs in a associated with HWS1 expression are arranged in the table (bottom), and the leading SNP variant is marked with a red arrowhead and asterisk. Hybrid combinations between SG178 and seven cultivars including four japonica varieties (d–g) and three indica varieties (h–j). The plants in parts d–j shown in the right panels exhibited typical sterile and dwarf phenotypes compared with the F2 control individuals shown on the left. Scale bars = 5 cm (top), 5 cm (bottom). k Allele frequencies of the causal polymorphisms in HWS1/HWS2 in the three groups comprising 133 AA-genome Oryza accessions. The total number and proportion of accessions in each class and population are given beneath each pie chart. The blank grid indicates that there were no varieties in this Class. All rice accessions are listed in Supplementary Data 1. Data in b are means ± SEM (n = 3 biological replicates). Different letters denote significant differences (p < 0.05, one-way ANOVA with two-sided Tukey’s HSD test). P values for b are adjusted and shown in the Source Data file.
On the basis of the above results, a presumptive model interpreting the duplication and ILS history of OsEAF6 in Oryza genomes was proposed as follows (Fig. 6). The ancestor EAF6 gene that originated from chromosome 2 of L. perrieri was inherited to chromosome 2 in the BB, CC, FF and GG genome species of Oryza, and then anchored on chromosome 1 in rice species with AA genome, possibly by transposon-mediated gene replication. This single-copy form of OsEAF6 gene on chromosome 1 was subsequently retained in the O.longistaminata, O.meridionalis, and O.glumipatula while an interchromosomal copy from chromosome 1 to chromosome 12 was produced in O.rufipogon followed by another ILS event occurred between O.barthii and O.rufipogon (Fig. 6a). In the primitive ancestor gene pool of O.rufipogon and O.barthii, there existed two major polymorphic HWS1 alleles: O.barthii-like (OB-like; oval) and O.rufipogon-like (OR-like; rectangle). Unlike that only OB-like HWS1 allele was inherited and directed fixed in O.barthii, these two types of HWS1 alleles were both preserved in the ancestor of O.rufipogon due to the ILS event. Then, a duplication event that generated the HWS2 allele replicated from the OR-like HWS1 and formed the Class I-type O.rufipogon. The Class I-type O.rufipogon was next crossed with the OB-like wild rice ancestor and formed the heterozygous Class I-type O.rufipogon. A random T to C mutation at the promoter region of the OB-like HWS1 allele was then initiated in the heterozygous Class I-type O.rufipogon. This T/C variation in the heterozygous Class I-type O.rufipogon eventually gave rise to the form of Class I/II/III-type O.rufipogon in the hybrid progenies (Fig. 6b). The various alleles of the O.rufipogon accessions eventually evolved to the Class I-III of O.sativa and the O.barthii, ancestor of African rice, evolved to the Class II type of O.glaberrima (Fig. 6).

a a simplified schematic diagram illustrating the evolutionary trajectory of the OsEAF6 genes after gene duplication and ILS events in AA and non-AA genome Oryza species. The currently existing OsEAF6 alleles in the Oryza genus trace back to the progenitor singleton on chromosome 2 of L. perrieri and evolved into chromosome 2 of BB, CC, FF, and GG genome rice. Finally, the OsEAF6 gene replicated to chromosome 1 of the ancestor AA genome rice. The orange and green lines indicate the evolution and ILS events, and the black arrows indicate the duplication event. The numbers 1, 2, and 12 in each oval represent the chromosomes on which the copied EAF6 genes are located. b a diagram showing the origin and generating process of Class I-III of HWS1/2 combination in O.barthii and O.rufipogon. Two HWS1 types, O.barthii-like (OB-like; oval and light green) and O.rufipogon-like (OR-like; rectangle and dark green) alleles, co-existed in the ancestral gene pool of O.rufipogon and O.barthii while HWS2 was generated later by duplication from the OR-like HWS1, leading to the generation of Class I-type O.rufipogon. By crossing with the OB-like ancestor wild rice, the heterozygous Class I-type O.rufipogon was subsequently formed. The produced T-to-C mutation in the promoter region of OB-like HWS1 allele occurred in the heterozygous Class I-type of O.rufipogon. The heterozygous Class I-type of O.rufipogon carrying the T/C variation in the OB-like HWS1 eventually produced the Class I-III of O.rufipogon in the self-progeny. mRNA transcripts of HWS1 and HWS2 are marked with the characters “+”, and “−” in rectangles and ovals to indicate the transcriptional products with high accumulation (HWS1, dark and light green; HWS2, purple) or transcripts that are expressed at low levels (hws1, gray) based on the speculative causative mutation. Additionally, the absence of HWS2 is signified by dashed lines, and the hybrid HWS1/2 is indicated with the solid and dashed lines.
