

Abstract
Assay for Transposase Accessible Chromatin by sequencing (ATAC-seq) accurately depicts the chromatin regulatory state and altered mechanisms guiding gene expression in disease. However, bulk sequencing entangles information from different cell types and obscures cellular heterogeneity. To address this, we developed Cellformer, a deep learning method that deconvolutes bulk ATAC-seq into cell type-specific expression across the whole genome. Cellformer enables cost-effective cell type-specific open chromatin profiling in large cohorts. Applied to 191 bulk samples from 3 brain regions, Cellformer identifies cell type-specific gene regulatory mechanisms involved in resilience to Alzheimer’s disease, an uncommon group of cognitively healthy individuals that harbor a high pathological load of Alzheimer’s disease. Cell type-resolved chromatin profiling unveils cell type-specific pathways and nominates potential epigenetic mediators underlying resilience that may illuminate therapeutic opportunities to limit the cognitive impact of the disease. Cellformer is freely available to facilitate future investigations using high-throughput bulk ATAC-seq data.
Similar content being viewed by others


Introduction
Transcriptional regulation and chromatin accessibility have been shown to play a crucial role in various neurological disorders1. Among other epigenetic techniques, the Assay for Transposase Accessible Chromatin by sequencing (ATAC-seq) provides an accurate way to depict the chromatin landscape of the brain and how it is altered by neurodegenerative diseases2,3,4. ATAC-seq is notably relevant to nominate candidates involved in disease, especially non-coding regions that disrupt gene transcription. While bulk ATAC-seq promises to determine open chromatin regions (OCR) and gene regulatory changes in a direct and efficient way, it entangles data from different cell types and obscures cell type-specific information5,6,7. Although single nucleus (sn) ATAC-seq can overcome this barrier7, it is labor-intensive, expensive, and vulnerable to technical dropout impacting data analysis and interpretation8.
Deconvoluting bulk sequencing data has been widely investigated, especially for RNA-sequencing9,10,11,12,13,14 and recently adapted for bulk ATAC-seq as OCR is better at capturing cell type-specificity than gene expression12,15,16,17. These computational approaches rely on a well-designed signature matrix, using only limited and most distinguishing cell type-specific features to estimate the cellular composition of tissue samples. While this matrix approach can help to resolve spatial single-cell gene expression from bulk RNA-seq18, the definition of cell type-specific marker remains challenging14,19. Recently, a deep learning approach has been proposed to bypass this limitation and directly predict cellular abundance from bulk RNA and microarray expression with high accuracy13. Cellular abundance change is a milestone in bulk analysis and has led to new insight into biological mechanisms13,20. Yet, it prevents a comprehensive understanding of the chromatin accessibility heterogeneity across cell populations and cell-specific OCR variation in disease, limiting bulk sequencing analysis.
Source separation21 is a widely studied signal processing paradigm that retrieves the set of individual sources from a mixed signal. One classical application is in audio processing, where one microphone is recording multiple instruments that are playing simultaneously. Source separation consists in retrieving the sound made by each type of instrument individually from one recorded audio signal. In this study, using a similar paradigm, we develop and test a deep learning-based algorithm, Cellformer, that separates the expression of 6 main brain cell types from bulk samples: 4 glial cell types including astrocytes (AST), microglia (MIC), oligodendrocytes (OLD) and oligodendrocyte progenitor cells (OPCs) and 2 major classes of neurons, excitatory (EXC) and inhibitory (INH). Unlike previous studies, Cellformer not only estimates cellular composition but also deconvolutes cell type-specific ATAC-seq OCR along the whole genome.
As we age past 65 years, the majority of the population resides on the Alzheimer’s disease (AD) continuum, meaning that approximately four out of five older adults have latent, prodromal, or full expression of AD dementia (ADD)22,23. These stages within the AD continuum typically have a progression of functional decline matched with increasing disease burden as measured during life by histopathology, neuroimaging, or biomarkers24,25,26. Standing apart from the AD continuum is a relatively small subset of older individuals who have mismatched normal cognitive function and a high disease burden sufficient to cause dementia; these individuals, called resilient to AD (RAD), are especially important because their existence demonstrates that even advanced AD burden does not necessarily lead to dementia. What combination of inherited factors, life choices, and experiences incurred or avoided combine in this “natural protection” that can be fully effective even in centenarians? Recently, several putative genetic loci involved in RAD have been found using genome-wide association studies (GWAS)27,28, yet the underlying gene regulatory machinery that mediates gene expression in RAD remains to be elucidated.
In this work, we leverage 191 well-curated tissue samples from sex and age range matched normal control (NC, n = 5), RAD (n = 12), and ADD (n = 19) individuals, without neurological comorbidities, and use Cellformer to predict cell type-specific ATAC-seq data from three brain regions and provide unique insights into the cellular and molecular mechanisms underpinning RAD (Fig. 1, Supplementary Fig. 1).

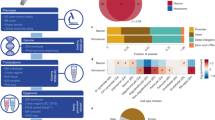
Cellformer was fed data from comorbidity-free bulk samples from individuals with clinicopathologic characterization as normal control (NC), Resilient to Alzheimer’s disease (RAD), and Alzheimer’s disease dementia (ADD). Three brain regions were used per individual to gain insight into the regional and cellular epigenetic profile of RAD. Cellformer generated cell type-specific expression for 6 main cell types across the whole genome, leading to an unprecedented chromatin profiling of RAD (Created with Biorender.com).
Results
Cellformer: from bulk to cell type-specific OCR
The rich diversity of cell type-specific changes can be obscured in bulk tissue transcriptomic and epigenomic analyses by mixing across heterogeneous cell populations. Hence, we hypothesize that deep learning algorithms, developed to separate mixed source signals21, could help resolve cell type-specific expression. However, a major pitfall of deep learning is the requirement of large and annotated datasets to train the model without overfitting, yet bulk ATAC-seq datasets with corresponding known cell type-specific expression compositions are very scarce. To bypass this limitation, we leveraged single-nucleus ATAC-seq collected from the brains of NC individuals7 and an in-silico dataset generation strategy to create synthetic bulk samples with established cell type-specific expression13 (Fig. 2a). More precisely, Cellformer was trained using synthetic subject-specific synthetic bulk samples. These samples were generated by first sampling and merging a random number of single nuclei per cell type, to create cell type-specific ground truth. Then combining cell type-specific expression produces synthetic bulk samples, Cellformer’s input (Methods).

a A synthetic dataset of simulated bulk samples was generated from previously published single-cell ATAC-seq from 13 normal controls7. Cell type-specific pseudo-bulk samples were generated by aggregating snATAC-seq data, revealing the ground truth cell type-specific composition. Simulated cell-specific pseudo-bulk samples were further aggregated to generate pseudo-bulk samples, which are Cellformer’s input. This dataset was used to train Cellformer to minimize the reconstruction error between predicted and ground truth cell type-specific ATAC-seq (Created with Biorender.com). b Cellformer leverages a dual-path strategy to process both intra and inter-chromosome interaction, enabling full genome deconvolution. P values were derived using a two-sided Wilcoxon’s test after multi-testing correction. c Cellformer was evaluated using the leave-one-subject-out strategy. It outperformed other multi-output regression models, notably linear regression, KNN and an unsupervised approach (NMF) used previously to estimate cellular composition across the (n = 6) different cell types. P values were derived using a two-sided Wilcoxon’s test after multi-testing correction. d Cellformer successfully deconvoluted leave-one-out cross-validated PBMC in-silico bulk ATAC-seq data from different datasets (n = 18 samples), predicting cell type-specific expression of five main cell types (B cell, T cell-CD4+ (CD4), T cell-CD8+ (CD8), Myeloid and NK cells). P values were derived using a two-sided Wilcoxon’s test after multi-testing correction. e Quality of the Cellformer’s predictions was assessed by comparing technical replicate cell type-specific expression (n = 36 samples, see Fig. 1). Cellformer generated outputs that are highly consistent between true technical replicates, exhibiting a correlation coefficient (>0.9) significantly higher than with random replicates. (Two-sided Wilcoxon’s test after multi-testing correction) f Cellformer output preserves cell type signature across 6 cell types: astrocytes (AST), microglia (MIC), oligodendrocytes (OLD), and oligodendrocyte progenitor cells (OPCs), and 2 major classes of neurons, excitatory (EXC) and inhibitory (INH). An external cell classifier trained on single-cell data from NC samples was used to assess the cell type-specific ATAC-seq quality. The confusion matrix computed between the cell classifier and Cellformer predictions showed almost perfect agreement, highlighting its capacity to preserve the cell type signature. g Cellformer validation was performed by comparing RAD cell type-specific expression from SMTG with RAD single-cell ATAC-seq expression from SEA-AD using a two-sided Spearman correlation. Significant high correlations were obtained within the same cell type between the two datasets. Spearman correlation coefficient order between cell types was consistent with biological knowledge: a high correlation was found between neuron types and between OLD and OPCs. All box plots show the median (middle line), interquartile range (bottom and upper edges), and the minimum and maximum values of the distribution (whiskers). *P value < 0.05, **P value < 0.01, ***P value < 0.001, ****P value < 0.0001.
Processing DNA-based sequencing has inherent challenges including handling the extremely large number of sequential features, which can lead to both memory and computational challenges. Deep learning offers the promise of dealing with high dimensional data and showing successful applications in diverse tasks using ATAC-seq data29,30. To handle the ATAC-seq high-dimensionality, Cellformer combines attention mechanisms and an effective method, named dual-path. The attention mechanisms create connections between distantly related elements demonstrating high performances in long-sequence modeling with the development of Transformer models8f).
To complement our analysis and nominate RAD gene regulatory elements, an activity-by-contact (ABC) algorithm was applied to the set of predictable OCR and HiChip from different brain regions7 to predict regional gene enhancer interactions47. ABC model determined 16,320 hippocampal enhancer OCR with 15% showing physical evidence only in this region (Supplementary Fig. 8g). By intersecting the set of predicted enhancers with RAD-specific OCR, we found that <50% of RAD-specific OCR were linked to cis-regulated elements while the rest were found in non-coding regions (Fig. 4e). GO and pathway analysis applied to OCR predicted to be localized in both genic and intergenic RAD-specific enhancers revealed significant enrichment of genes related to chemical synaptic transmission in excitatory neurons, inhibitory neurons, and microglia notably Amyloid Beta Precursor Protein Binding Family A Member 2 (APBA2), that modulates AD amyloid precursor protein, and BDNF signaling pathway. These findings corroborate previous analyses performed with microarray protein analysis and animal models48,49.
Discussion
Bulk ATAC-seq is an effective and efficient method to measure open chromatin accessibility2,4,35. In human brains, ATAC-seq may be favored over RNA-based methods for technical reasons, including greater stability of DNA in post-mortem brains and more comprehensive assessment than in single-nucleus assays50. While snATAC-seq offers the attractive opportunity to detect cell type-specific open chromatin accessibility, it is highly impacted by dropout events, making snATAC-seq analysis more challenging and vulnerable to missing low-expressed genes51. To remedy this, we developed Cellformer, a new approach to deconvolute bulk ATAC-seq data and thereby computationally enhance resolution to the cell-type level. Using Cellformer, we illustrated the power of deep learning to enhance biological data analysis and advance our understanding of disease mechanisms at the epigenetic level.
The cellular composition of tissue is a crucial component of sequencing analysis40,52. In contrast to previous methods to deconvolute bulk sequencing data9,12,53, Cellformer does not aim to predict cellular abundance in bulk tissue but instead “fully” deconvolutes bulk ATAC-seq, generating OCR data at the cell type level across the whole genome. Besides, Cellformer does not rely on cell type signature matrix definition9,18, a key ingredient of previous bulk deconvolution that strongly correlates with prediction accuracy14,19 and deters application to samples where no single-cell data are available, such as the RAD and ADD samples investigated here. Defining an accurate signature remains an open computational problem that has been poorly investigated for ATAC-seq data12. Cellformer bypasses this issue by leveraging the power of deep learning to automatically extract and predict cell type-specific OCR13.
While ATAC-seq can be performed on experimentally isolated single cells or single nucleus, Cellformer can resolve bulk expression at the cell type level, not at the single-cell level. However, creating cell type-specific mixtures is a popular strategy to overcome the low count and sparsity of snATAC-seq and improves the statistical power of single-cell analysis54. Notably, cell type-specific data are used to strengthen the signal and improve statistical significance for high-confidence, differential analysis55, TF footprint, or disease gene regulatory signatures identification56,57,58. Furthermore, similar to single-cell sequencing, Cellformer is limited to the most predictable and highly expressed cell type-specific OCR.
Cellformer belongs to the reference-based method category: supervision of the model requires snATAC-seq to learn cell type-specific expression. Therefore, Cellformer predictions are limited to the cell type-specific open chromatin patterns detected in the single-nucleus samples and strongly depend on the quality of snATAC-seq samples.
In this study, we focused on the 6 major brain cell classes. Although we demonstrate Cellformer’s ability to deconvolute at a lower resolution (Supplementary Fig. 4b), we notice that increasing the number of output cell types results in a significant rise in computational complexity. This limitation restricts the number of deconvoluted cell types that can be effectively handled. To overcome this challenge, we plan to implement and validate strategies such as hierarchical training or cell-type prioritization functions, which will expand the capabilities of Cellformer. The model generalization and robustness will be also improved in the future by leveraging the ever-growing amount of available single-cell/nucleus ATAC-seq data or by adapting transfer learning approaches to improve prediction performances on bulk samples from new tissues. Another limitation is the time of training which varies between two hours and a few days depending on the model configuration, the computing power used for the training, and the number of samples. Yet, it remains faster and less expensive than a single-cell sequencing protocol. Additional hyperparameter optimization and gradient acceleration strategy will be implemented in the future to improve training efficiency59.
Applied to large cohort bulk ATAC-seq data from multiple human brain regions, Cellformer provided new insights into RAD, an unusual group of individuals who do not succumb to the high burden of AD and who likely hold important clues to treat this highly prevalent disease. Cellformer predicted that OCR differences between RAD and the other two groups that are on the AD continuum were very strongly localized to HIPP, which subserves declarative memory formation and is the primary target of AD. At the cellular level, most predicted RAD-specific OCR were characterized by changes in both inhibitory and excitatory neurons followed by microglia60. From the perspective of cellular processes, GO analysis of RAD-specific OCR highlighted neuronal development, inflammatory response, and synaptic transmission processes. These pathways were highlighted in previous studies using proteomics and mouse models of AD48,61,62,63. Overall, these highly plausible predictions suggest that individuals with RAD are distinguished from the AD continuum by epigenetic upregulation in support of hippocampal neuronal processes and synapses. This regulation change might confer RAD the ability to preserve the number of neuronal projections and synapses that have been observed through histopathological studies64.
Cellformer offers new perspectives to gain insight into bulk sequencing and identify cell-specific gene regulatory changes in disease progression in a cost-effective way. Complementing cellular abundance prediction, Cellformer provides additional information to advance bulk ATAC-seq analysis. We expect that Cellformer may help to unveil cell-specific transcriptional regulation and advance our understanding of disease epigenetic mechanisms in other biological settings.
Methods
Data overview
This study drew on snATAC-seq and bulk ATAC-seq data previously collected7. Primary brain samples were obtained from Stanford University, the University of Washington, or Banner Health from post-mortem tissue following informed consent and Institutional Review Boards approval7 (Supplementary Fig. 1). In addition, five recently collected control snATAC-seq samples from SMTG (collected using the same ATAC-seq protocol7) were used to train the model. Data on sex was collected but not gender. Validation was performed using Seattle Alzheimer’s Disease Brain Cell Atlas (SEA-AD)43 single-cell (sc-) ATAC-seq data generated using a 10xMultiome preparation, snATAC-seq from human prefrontal cortex42 and genetic variants from the most recent GWAS study for late-onset Alzheimer’s45.
Individuals’ brain samples from both ATAC-seq datasets were carefully filtered according to clinical diagnosis of cognitive status proximate to death and assessment of AD neuropathologic change and other neuropathologic comorbidities (see Supplementary Table 1) using current consensus guidelines24,65,66,67,68,69. Resilient cases were defined as individuals without dementia at their most recent clinical research evaluation within 2 years of death, and neuropathologic findings of B score >2 and C score >1 but without vascular brain injury or Lewy body disease, and LATE neuropathologic change stage of 0 or 1. To ensure a valid comparison between cell-type-specific ATAC-seq and single-nucleus ATAC-seq from SEA-AD cohort, we also removed samples from patients with known Lewy Body disease (brainstem, limbic, neocortical, olfactory) and LATE neuropathologic change stage above 1 in the SEA-AD dataset.
Bulk ATAC-seq and scATAC-seq processing
We leveraged annotated single-cell ATAC-seq from 12 NC subjects to identify cell type-specific OCR (or peak)7. “Peak calling” was performed on regional and cell type-specific replicates to improve statistical significance using ArchR workflow56 and its MACS2 implementation70. Chromatin accessibility varies largely per brain region and cell type1. Therefore, to ensure capturing OCR that is significantly expressed in the 6 main cell types of interest (astrocyte, microglia, oligodendrocyte, OPCs, excitatory, and inhibitory neurons), we removed single cells that do not belong to one of these six categories as well as single cells identified as doublets. Only the significant OCR marker56, unique to an individual or a small number of cell type groups was conserved for downstream analysis (FDR < 0.001 and FC > 1). In total, we defined a set of 41,954 OCR. The count normalized matrix, combining OCR from all the bulk samples, was then derived using featureCount71.
Previously preprocessed and annotated PBMC scATAC-seq was downloaded from (https://github.com/GreenleafLab/ArchR_2020)56. Peak calling, peak filtering, and in-silico bulk generation were performed using the same workflow as described above.
FeatureCount (version 2.0.3) also was applied to 10xMultiple single-nucleus ATAC-seq fragment files from SEA-AD cohort to extract the same set of OCR. For an accurate comparison, we only considered predictable cell type-specific OCR for comparison, used in this study. Only cells passing the quality control and annotated using the paired snRNA were used for downstream analysis43.
Annotated raw OCR count matrix provided by Morabito et al42. was used and intersected with our set of predictable OCR to compare the prefrontal cortex from ADD and NC single nucleus with deconvoluted cell type expression using “intersect” function from bedtools72. A set of 20060 overlap** OCR was then used to compute the Spearman correlation matrices between cell type expression (Supplementary Fig. 8).
Transformer-based cell-specific ATAC-seq separator
The “Cocktail party” or source separation problem is a widely studied question consisting of extracting individual source signals from a mixed one. Inspired by this paradigm, we leveraged a state-of-the-art source separation method to deconvolute bulk ATAC-seq and extract individual cell type-specific expression along the whole genome. Comprehensive processing of the gene regulatory elements linkage, spanning the whole genome, was achieved using a long-sequence friendly neural network developed for speech separation34. Based on the inner/outer transformer-based architecture, our neural network can extract both within and cross-chromosome epigenetic dependencies along the whole genome using a reduced number of trainable parameters. More precisely, this “dual-path recurrent neural network” strategy73,74 decomposes long sequences into smaller chunks of size 250 and, extracts high-level representations within chunks, which are then concatenated and permuted for inter-chunk interaction processing. Dual path-based models have shown to be effective at modeling very long sequences, leading to superior predictive performances in various audio processing tasks. We adapted the published model to predict the ATAC-seq profile of 6 major brain cells including astrocytes, microglia, oligodendrocytes, OPCS, and two subclasses of neurons, excitatory and inhibitory neurons. An advantage of this architecture is that it can extract both within and cross-chromosome cell type-specific epigenetic dependencies all along the genome.
To improve our prediction confidence and enhance the robustness of our approach, we stacked on top of our trained network a filtering module removing predicted OCR with a relatively high training error. For each cell type i, we computed the normalized mean absolute error across all the N samples in the training set as: \({{{{{\rm{NMEA}}}}}}_{p}=\frac{{{{{\rm{MEA}}}}}({\hat{{{{{{\bf{X}}}}}}}}_{i},{{{{{{\bf{X}}}}}}}_{i})}{\frac{1}{N}{\sum }_{p=1}^{N}|{{{{{{\bf{X}}}}}}}_{i}|}\), with \({{{{{{\bf{X}}}}}}}_{i}\) and \({\hat{{{{{{\bf{X}}}}}}}}_{i}\) the ground truth and the predicted, OCR values, respectively for the cell type i. We preserved cell type-specific OCR for downstream analysis with a mean error lower than a threshold defined as: \({{{{{\rm{NMEA}}}}}}_{p} \, < \, {M}_{{{{{\rm{NMEA}}}}}}+\tau*{\sigma }_{{{{{\rm{NMEA}}}}}}\), with \({M}_{{{{{\rm{NMEA}}}}}}\) and \({\sigma }_{{{{{\rm{NMEA}}}}}}\) the mean and standard deviation of NMEA across all the OCR and \(\tau\) the threshold ranging from −0.5 to 1. \(\tau\) was optimized using samples from the training to maximize the correlation between the cell type-specific ground truth and predicted ATAC-seq expression. Once the filtering module was trained, it was applied to the model outputs to preserve only the most predictable cell type-specific OCR.
The pythonic implementation from Asteroid librarySynthetic dataset generation Model training was achieved by creating a synthetic dataset of paired bulk and corresponding cell type-specific samples (ground truth), leveraging available single cells samples from NC12,13 Each pair in the synthetic dataset was created by first sampling and aggregating a random number of the same type of single nuclei from a sample’s snATAC-seq results in order to create synthetic cell type-specific pseudo-bulk samples that preserve regional and individual diversity in our synthetic dataset. Then, corresponding synthetic bulk ATAC-seq data were created by aggregating the generated cell type-specific pseudo-bulk samples from the six cell types. 3000 pairs of synthetic bulk and cell type-specific bulk were generated from each subject, composed of a random number of cells ranging from 100 to 800. Then, both synthetic cell type-specific and bulk samples were normalized by the total number of cells and the maximum OCR value. Harmony batch normalization was applied on snATAC-seq to ensure robust peak calling7,56. No batch normalization was further applied on the input of the model, since additional analysis suggests that Cellformer removes batch effects while preserving biological variations, i.e., kBet = 0.79 ± 0.14, NMI = 0, ARI = −0.02 ± 0.001, cLISI = 1, iLISI = 0.58 ± 0.02 across the brain regions77. Model generalizability was assessed using the leave-one-subject-out-strategy. More precisely, at each iteration, the dataset was partitioned into a training and testing set, such that samples from all brain regions from one individual were left out while the rest of the samples were gathered to create the training set. From the training set, 20% of the samples were used to tune the model hyperparameters at each iteration while the remaining samples helped to optimize the weights of the model. Once trained, we fed the model with synthetic bulk samples created by aggregating single cells per snATAC-seq donor from the test set (never seen by the model) and validated its performances at predicting an accurate and consistent cell-specific signal using the Spearman correlation. We also assessed the ability of the model to predict non-zero OCR by computing the AUROC and AUPRC after binarization of the ATAC-seq expression29,30. Mean errors with quartile error bars across iterations were reported and compared with other models. The model with the highest performance on the whole dataset and among the top three models with the lowest test error was used for downstream analysis. Model output consistency was validated by computing the Spearman correlation between technical replicates and predicted cell type-specific signals (Fig. 2e). To assess the significance of the observed mean correlation between technical, random replicate permutation tests were performed. More precisely, for each bulk sample, Spearman correlation was computed between the model’s output of this sample and a random replicate, arbitrarily selected from the same brain region, from the phenotype group, or both the same brain region and phenotype group (Supplementary Fig. 3). P value was derived by comparing the mean correlation between true replicates and random replicates using Bonferroni corrected two-sided Wilcoxon test. We ensured the cell type signatures model’s preservation in ADD and RAD samples using an external cell classifier (Supplementary Fig. 6). To better capture the cell type-specific signature and be more robust to dropout, an XGBoost classifier was trained to classify single-nucleus ATAC-seq from Control cases into cell class. The model was trained to minimize a softmax loss between the predicted label from synthetic cell type-specific ATAC and the corresponding ground truth (see Synthetic Dataset Generation). Stratified K-fold nested cross-validation was exploited to validate the model and performances of the model were quantified using AUROC and AUPRC between the ground truth label and the predicted probability and optimized the model hyperparameters. The XGBoost package in Python was exploited to implement the cell classifier, parameterized with a learning rate equal to 0.1, a maximum depth of tree set to 10 and 100 estimators. The model achieved a mean AUROC = 1.000, Precision = 0.994, and Recall = 0.993 over cross-validation iterations when tested on the held-out single-nucleus ATAC-seq datasets. Once trained and validated to accurately predict the cell type, the model was applied to deconvoluted cell type-specific expression from AD and RAD. The classifier achieved an AUROC of 1.000, a Precision of 0.993, and a Recall of 0.994 when tested on the deconvoluted expression. Confusion matrices showed almost a perfect classification across cell types, brain regions, and conditions. We compared our model with supervised (Linear regression), nonparametric (KNN) machine learning, and unsupervised (Non-Negative Matrix Factorization) models. These algorithms were implemented using the default parameterized functions from Scikit-learn78. Using 100 synthetic bulk samples, the multi-out models (Linear regression and KNN) were trained to predict cell type-specific OCR by minimizing the MSE loss. The same leave-one-subject-out strategy as for Cellformer was used to assess models’ generalization and avoid overfitting. NMF model was trained using a synthetic bulk matrix created by aggregating all the single nuclei per replicate. Then, the predicted OCR-specific expression was computed through row-wise multiplications between the feature matrix and the coefficient vectors. Chipseeker79 was used to identify OCR-gene association and genomic OCR annotation using default parameters following ATAC-seq data processing guidelines and Harvard bioinformatics recommendations35,80. One of the main issues in epigenetic analysis is the lack of consensus between annotating tools81. We, therefore, compared Chipseeker to the annotations given by ArchR, developed for ATAC-seq data analysis. Overall, Chipseeker and ArchR agreed on 60% of peaks, including complete (100%) agreement between peak-to-gene annotations of RAD-specific promoters, from which our biological insights were drawn. In particular, similar gene ontology enrichment is observed using both tools. Candidate enhancer OCRs per brain region were computed using Active-by-Contact (ABC) model47. Candidate enhancer regions were derived for each brain region independently using 10 ATAC-seq replicates. Region-specific ABC scores were computed by combining the OCR activity and the genomic spatial information extracted from the HiChip-seq data provided by Corces et al7., using the suggested parameters80. Differentially expressed OCR were identified using FDR corrected two-sided Wilcoxon test using FDR 5% and absolute log fold-change superior to 0.5 from Scanpy library and MultiPy82,83,84. Gene ontology and pathway analysis were performed on OCR-related genes with the GO and BioPlanet databases from 2021 and 2019, respectively, using GSEAPY85 (version 1.0.3). Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.Model validation
Model testing through leave-one-subject-out cross-validation
Model’s output consistency and plausibility
Cell signature preservation
Model comparison
OCR annotation
Cell type-specific ATAC-seq analysis
Reporting summary
Data availability
All data supporting the findings described in this manuscript are publicly available. Bulk ATAC-seq and single-cell ATAC-seq from control individuals were previously collected and annotated26 accessible through GEO accession (GSE147672). Additional single-nucleus ATAC-seq data and raw and processed bulk ATAC-seq from ADD and RAD are available through GEO accession (GSE226529) and Dryad (https://doi.org/10.5061/dryad.2fqz612t0). Validation of the model was performed using processed snATAC-seq from40,41 available at (http://portal.brain-map.org/explore/seattle-alzheimers-disease and https://www.synapse.org/#!Synapse:syn22079621/wiki/603535. Processed PBMC ATAC-seq data are accessible at https://github.com/GreenleafLab/ArchR_2020).
Code availability
For future research, all custom code used in this work code, processed data, and additional metadata have been made publicly available at (https://github.com/elo-nsrb/Cellformer) and https://doi.org/10.5281/zenodo.8175353. The following packages were used: Python 3 (version 3.9.7) with PyTorch (version 1.10.0); Scikit-learn (version 1.0.1), asteroid (0.5.2), and GSEAPY (version 1.0.3); R (version 4.2.2) with ArchR (R version 4.2.2), Chipseeker (version 1.36.0); FeatureCount (version 2.0.3).
References
Fullard, J. F. et al. An atlas of chromatin accessibility in the adult human brain. Genome Res. 28, 1243–1252 (2018).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Satpathy, A. T. et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936 (2019).
Grandi, F. C., Modi, H., Kampman, L. & Corces, M. R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 17, 1518–1552 (2022).
Novikova, G. et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 12, 1610 (2021).
Barker, S. J. et al. MEF2 is a key regulator of cognitive potential and confers resilience to neurodegeneration. Sci. Transl. Med. 13, eabd7695 (2021).
Corces, M. R. et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 52, 1158–1168 (2020).
Qiu, P. Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun. 11, 1169 (2020).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Cobos, F. A., Vandesompele, J., Mestdagh, P. & De Preter, K. Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 34, 1969–1979 (2018).
Frishberg, A. et al. Cell composition analysis of bulk genomics using single-cell data. Nat. Methods 16, 327–332 (2019).
Li, H. et al. DeconPeaker, a deconvolution model to identify cell types based on chromatin accessibility in ATAC-seq data of mixture samples. Front. Genet. 11, 392 (2020).
Menden, K. et al. Deep learning–based cell composition analysis from tissue expression profiles. Sci. Adv. 6, eaba2619 (2020).
Vallania, F. et al. Leveraging heterogeneity across multiple datasets increases cell-mixture deconvolution accuracy and reduces biological and technical biases. Nat. Commun. 9, 4735 (2018).
Song, L. et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 21, 1757–1767 (2011).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Corces, M. R. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016).
Liao, J. et al. De novo analysis of bulk RNA-seq data at spatially resolved single-cell resolution. Nat. Commun. 13, 6498 (2022).
Monaco, G. et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 26, 1627–1640.e7 (2019).
Luca, B. A. et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell 184, 5482–5496.e28 (2021).
Roweis, S. One Microphone Source Separation. In: Advances in Neural Information Processing Systems (eds. Leen, T., Dietterich, T. & Tresp, V.) vol. 13 (MIT Press, 2000).
Sonnen, J. A. et al. Ecology of the aging human brain. Arch. Neurol. 68, 1049–1056 (2011).
White, L. R. et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia aging studies. Neurology 86, 1000–1008 (2016).
Hyman, B. T. et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13 (2012).
Therriault, J. et al. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat. Aging 2, 526–535 (2022).
Jack, C. R. Jr et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018).
Dumitrescu, L. et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain 143, 2561–2575 (2020).
White, C. C. et al. Identification of genes associated with dissociation of cognitive performance and neuropathological burden: Multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med. 14, e1002287 (2017).
Wu, K. E., Yost, K. E., Chang, H. Y. & Zou, J. BABEL enables cross-modality translation between multiomic profiles at single-cell resolution. Proc. Natl. Acad. Sci. USA 118, e2023070118 (2021).
Lal, A. et al. Deep learning-based enhancement of epigenomics data with AtacWorks. Nat. Commun. 12, 1507 (2021).
Vaswani, A. et al. Attention is all you need. Ar**v https://arxiv.org/abs/1706.03762 (2017).
Karita, S. et al. A comparative study on transformer vs RNN in speech applications. In: 2019 IEEE Automatic Speech Recognition and Understanding Workshop (ASRU) https://arxiv.org/abs/1909.06317 (2019).
Le, N. Q. K. & Ho, Q.-T. Deep transformers and convolutional neural network in identifying DNA N6-methyladenine sites in cross-species genomes. Methods 204, 199–206 (2022).
Subakan, C., Ravanelli, M., Cornell, S., Bronzi, M. & Zhong, J. Attention is All You Need in Speech Separation. ICASSP 2021-2021 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP). 21–25 (2021).
Yan, F., Powell, D. R., Curtis, D. J. & Wong, N. C. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biology 21, 22 (2020).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 11, 34 (2019).
Racle, J., de Jonge, K., Baumgaertner, P., Speiser, D. E. & Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 6, e26476 (2017).
Chang, W. et al. ICTD: A semi-supervised cell type identification and deconvolution method for multi-omics data. bioRxiv https://www.biorxiv.org/content/10.1101/426593v3.full (2019).
Liu, Y. scDeconv: an R package to deconvolve bulk DNA methylation data with scRNA-seq data and paired bulk RNA-DNA methylation data. Brief Bioinform. 23, bbac150 (2022).
Chu, T., Wang, Z., Pe’er, D. & Danko, C. G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 3, 505–517 (2022).
Granja, J. M. et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 37, 1458–1465 (2019).
Morabito, S. et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet. 53, 1143–1155 (2021).
Travaglini, K. J. et al. A multimodal atlas of the molecular and cellular changes to cortex driven by Alzheimer’s disease. Alzheimers Dement. 18, e060835 (2022).
BRAIN Initiative Cell Census Network (BICCN). A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 598, 86–102 (2021).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Merrihew, G. E. et al. A peptide-centric quantitative proteomics dataset for the phenotypic assessment of Alzheimer’s disease. Sci. Data 10, 206 (2023).
Fulco, C. P. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Arnold, S. E. et al. Cellular, synaptic and biochemical features of resilient cognition in Alzheimer’s disease. Neurobiol. Aging 34, 157 (2013).
Nagahara, A. H. et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med 15, 331–337 (2009).
Rocks, D. et al. Cell type-specific chromatin accessibility analysis in the mouse and human brain. Epigenetics 17, 202–219 (2022).
Barrett, A. et al. Integrating bulk and single cell RNA-seq refines transcriptomic profiles of specific C. elegans neurons. bioRxiv https://www.biorxiv.org/content/10.1101/2022.04.05.487209v1 (2022).
Kuhn, A., Thu, D., Waldvogel, H. J., Faull, R. L. M. & Luthi-Carter, R. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat. Methods 8, 945–947 (2011).
Avila Cobos, F., Alquicira-Hernandez, J., Powell, J. E., Mestdagh, P. & De Preter, K. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun. 11, 5650 (2020).
Baek, S. & Lee, I. Single-cell ATAC sequencing analysis: from data preprocessing to hypothesis generation. Comput. Struct. Biotechnol. J. 18, 1429–1439 (2020).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Pliner, H. A. et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol. Cell 71, 858–871.e8 (2018).
Li, Z. et al. Chromatin-accessibility estimation from single-cell ATAC data with scOpen. Nat. Commun. 12, 6386 (2021).
Tan, M. & Le, Q. EfficientNetV2: smaller models and faster training. In: Proceedings of the 38th International Conference on Machine Learning (eds. Meila, M. & Zhang, T.) 139, 10096–10106 (PMLR, 2021).
Duggan, M. R. & Parikh, V. Microglia and modifiable life factors: potential contributions to cognitive resilience in aging. Behav. Brain Res 405, 113207 (2021).
Kaczorowski, C. C., Sametsky, E., Shah, S., Vassar, R. & Disterhoft, J. F. Mechanisms underlying basal and learning-related intrinsic excitability in a mouse model of Alzheimer’s disease. Neurobiol. Aging 32, 1452–1465 (2011).
Neuner, S. M. et al. Translational approaches to understanding resilience to Alzheimer’s disease. Trends Neurosci. 45, 369–383 (2022).
Seto, M., Weiner, R. L., Dumitrescu, L. & Hohman, T. J. Protective genes and pathways in Alzheimer’s disease: moving towards precision interventions. Mol. Neurodegener. 16, 1–16 (2021).
Perez-Nievas, B. G. et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526 (2013).
Montine, T. J. et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11 (2012).
Braak, H. & Braak, E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1, 213–216 (1991).
Nelson, P. T. et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527 (2019).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Mirra, S. S. et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486 (1991).
Zhang, Y. et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Chen, J., Mao, Q. & Liu, D. Dual-path transformer network: direct context-aware modeling for end-to-end monaural speech separation. Proc. Interspeech 2020. 2642–2646 (2020).
Luo, Y., Chen, Z. & Yoshioka, T. Dual-Path RNN: efficient long sequence modeling for time-domain single-channel speech separation. In: ICASSP 2020 − 2020 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP) 46–50 (IEEE). https://doi.org/10.1109/ICASSP40776.2020.9054266. (2020).
Pariente, M. et al. Asteroid: the PyTorch-based audio source separation toolkit for researchers. Ar**v https://arxiv.org/abs/2005.04132 (2020).
Paszke, A. et al. Pytorch: an imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 32, https://arxiv.org/abs/1912.01703 (2019).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2021).
Kramer, O. Scikit-Learn. in Machine Learning for Evolution Strategies (ed. Kramer, O.) 45–53 (Springer International Publishing, 2016).
Wang, Q. et al. Exploring epigenomic datasets by ChIPseeker. Curr. Protoc. 2, e585 (2022).
Gaspar, J. M. ATAC-seq guidelines. Harvard FAS Informatics. https://informatics.fas.harvard.edu/atac-seq-guidelines.html (2019).
Kondili, M. et al. UROPA: a tool for universal RObust peak annotation. Sci. Rep. 7, 1–12 (2017).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. 100, 9440–9445 (2003).
Puoliväli, T., Palva, S. & Palva, J. M. Influence of multiple hypothesis testing on reproducibility in neuroimaging research: a simulation study and Python-based software. J. Neurosci. Methods 337, 108654 (2020).
Fang, Z., Liu, X. & Peltz, G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757 (2023).
Acknowledgements
This work was supported by RF1 AG053959 (T.J.M.) U19 AG065156 (T.J.M.) R35GM138353 (N.A.), RF1 AG077443 (T.J.M., N.A.) U01 AG072573 (T.J.M.), NIH RM1-HG00735 (H.Y.C.). H.Y.C. is an Investigator at the Howard Hughes Medical Institute. Schematics were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
E.B., E.J.F., M.R.C., N.A., and T.J.M. conceived and designed the experiments. T.J.M., N.A., M.R.C., E.J.F., K.S.M., and H.Y.C. contributed to the funding acquisition. Samples were acquired by T.J.M., M.R.C., and H.Y.C., E.B. and A.S. performed the experiments with the help of T.P., T.J.M., N.A., M.R.C., A.P., N.G.R., L.X., N.P., S.M., Y.K., C.E. and A.L.C. E.B., A.S., A.P., F.G., L.X., N.G.R., N.P., S.M., Y.K., C.E., A.L.C., and M.B. contributed to data processing and analysis, visualization and analysis tools. E.B., N.A., K.S.M., and T.J.M. wrote the manuscript with the help of all the authors. All cohorts contributed to the manuscript review and editing.
Corresponding author
Ethics declarations
Competing interests
H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, Cartography Biosciences, and Orbital Therapeutics, and is an advisor of 10x Genomics, Arsenal Biosciences, Chroma Medicine, and Spring Discovery. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Berson, E., Sreenivas, A., Phongpreecha, T. et al. Whole genome deconvolution unveils Alzheimer’s resilient epigenetic signature. Nat Commun 14, 4947 (2023). https://doi.org/10.1038/s41467-023-40611-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40611-4
- Springer Nature Limited