

Abstract
Acid sphingomyelinase (ASM) has been implicated in neurodegenerative disease pathology, including Alzheimer’s disease (AD). However, the specific role of plasma ASM in promoting these pathologies is poorly understood. Herein, we explore plasma ASM as a circulating factor that accelerates neuropathological features in AD by exposing young APP/PS1 mice to the blood of mice overexpressing ASM, through parabiotic surgery. Elevated plasma ASM was found to enhance several neuropathological features in the young APP/PS1 mice by mediating the differentiation of blood-derived, pathogenic Th17 cells. Antibody-based immunotherapy targeting plasma ASM showed efficient inhibition of ASM activity in the blood of APP/PS1 mice and, interestingly, led to prophylactic effects on neuropathological features by suppressing pathogenic Th17 cells. Our data reveals insights into the potential pathogenic mechanisms underlying AD and highlights ASM-targeting immunotherapy as a potential strategy for further investigation.
Similar content being viewed by others


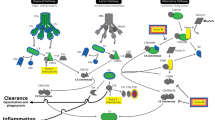
Introduction
Extensive research has shown the importance of circulating factors in the pathogenesis of aging and age-related neurodegenerative diseases such as Alzheimer’s disease (AD)1,2,3,4,5,6. For example, animal studies using heterochronic parabiosis, which is a technique combining the blood circulation of two animals, have revealed the powerful rejuvenating effects of young blood on aging and/or the age-related neurodegenerative brain disease7,8,54. However, limited amounts of circulating antibodies cross the BBB and enter the brain, and there is some uncertainty as to whether the positive therapeutic effects are due to antibodies that have reached the brain or are still present in the blood. Therefore, these studies remain somewhat controversial. In principle, based on our studies an immunotherapy targeting plasma ASM could overcome the limitation of crossing the BBB since its primary site of action would remain in the blood, not the brain tissue. In support of this concept, the results from parabiosis experiments using Smpd1−/− mice and active or passive immunotherapy in AD mice showed efficient inhibition of plasma ASM activity by ASM antibodies. Furthermore, we show marked prophylactic effects of the ASM antibody on neuropathological changes in AD despite no decrease of ASM activity in the brain. These results highlight the potential of this immunotherapeutic approach targeting plasma ASM for AD.
Notably, the inhibition of plasma ASM activity by the anti-ASM antibody also suppressed the pathogenic differentiation of Th17 cells in the blood, and contributed to normalization of other immune cell populations that affect brain neuroinflammation. This impact on pathogenic Th17 cells and other immune cell alterations is likely to contribute to the prevention of BBB disruption, neuroinflammation, Aβ accumulation, synapse loss, and even learning and memory impairment in the AD mice we observed (Supplementary Fig. 13). Thus, our findings suggested that plasma ASM-targeted immunotherapy could be a next generation immunotherapy for AD and, importantly, unlike other immunotherapies, does not require BBB penetration. Although we focused on the prophylactic effects of immunotherapy targeting plasma ASM in this study, we think that the ASM antibody also may have therapeutic effects in ameliorating neuropathological features in advanced AD mice as well. For this, we will be conducting further studies in the future. Elevated activity of plasma ASM is found in a variety of diseases including heart disease, diabetes mellitus, and inflammatory diseases such as sepsis and systemic inflammatory response syndrome19. If the pathological roles of plasma ASM is revealed in these diseases, immunotherapy targeting plasma ASM could be useful in these diseases as well.
Methods
Mice
The following mouse lines were used: C57BL/6 wild type (WT) mice (The Jackson Laboratory), Tie2-Cre mice (stock number 008863, The Jackson Laboratory), loxP-flanked Tg-Smpdstop mice22 (Smpd1ox/ox mice, C57BL/6 background), Smpd1−/− mice33 (C57BL/6 background), and Il17a-EGFP knockin mice (stock number 018472, The Jackson Laboratory). To obtain endothelial cell-specific ASM overexpressing mice, Tg-Smpdstop mice were crossed with Tie2-Cre mice. Transgenic mouse lines overexpressing the hAPP695swe (APP) and presenilin-1M146V (PS1) mutations were originated from GlaxoSmithKline (Harlow, UK)25 and maintained as described previously18,21. We used littermate mice that were sex- and age-matched between experimental groups. Both male and female mice were used for all experiments except behavioral studies using male mice.
The block randomization method was used to allocate the animals to experimental groups. To eliminate the bias, all investigators were blinded to the experimental groups and analysis such as data collection and data analysis. Mice were housed at a 12 h day/12 h night cycle, 21–22 °C and 50–60% humidity with free access to water and food pellets. All protocols were approved by the Kyungpook National University Institutional Animal Care and Use Committee (IACUC).
Parabiosis and IL17 antibody treatment
Parabiosis surgery followed previously described procedures7,12. To investigate the effects of overexpressed plasma ASM on AD pathologies, 3-month-old of APP/PS1 and age- and weight-matched WT or Smpd1ox/ox (Tie2-cre; Smpd1ox/ox) mice were selected for parabiosis surgery. IgG Isotype control (50 μg/mouse, R&D system, MAB0006) or IL17 antibody (50 μg/mouse, R&D system, MAB421) was injected intraperitoneally into APP/PS1 mice in parabiosis with WT or Smpd1ox/ox mice every other day for 5 weeks during parabiosis. To confirm the effects of antibody-based plasma ASM inhibition on AD pathologies, 7.5-month-old of APP/PS1 and age- and weight-matched APP/PS1, WT, or Smpd1−/− mice were selected for parabiosis surgery. Mirror-image incisions at the left and right flanks of age- and weight-matched mice were made through the skin, and shorter incisions were made through the abdominal wall. The peritoneal openings of the adjacent parabionts were sutured together. Elbow and knee joints from each parabiont were sutured together, and the skin of each mouse was stapled (9-mm Autoclip, Clay Adams) to the skin of the adjacent parabiont. Each mouse was injected subcutaneously with Baytril antibiotic and monitored during recovery. Five weeks after surgery, mice were sacrificed for analysis.
Mouse plasma or serum collection
Mouse blood was collected into sodium heparin-coated tubes via intracardial bleed at the time of death. Plasma was generated by centrifugation (15,493 × g, 4 °C, 5 min) of freshly collected blood and aliquots were stored at −80 °C until use. To collect serum, mouse blood was collected in a e-tube and the blood was allowed to clot by leaving it undisturbed at room temperature for 30 min. Serum was collected by centrifugation (15,493 × g, 4 °C, 5 min) and aliquots were stored at −80 °C until use.
Human plasma collection
Human plasma samples were obtained from both men and women with AD and age-matched non-AD controls from Hanyang University Hospital (Supplementary Table 1). Sex of participants was determined based on self-report. Informed consent was obtained from all subjects according to the ethics committee guidelines at the Hanyang University Hospital (IRB no. HYUH 2016-12-029-003).
Preparation of brain microvessels
Brain microvessels were prepared as previously described17. To prepare the brain samples, mice were anesthetized and blood was collected at the time of death into EDTA-coated tubes via intracardial bleed. After blood collection, mice were transcardially perfused with PBS and brains were further removed from the skull. The meninges and choroid plexuses were removed, and the brainstem and cerebellum were dissected away from the cerebrum. Following dissections, tissues were rinsed, and diced into small pieces (approx. 1 mm). Each tissue was then homogenized in Dounce tissue grinder. Resulting homogenates were centrifuged at 1000 × g for 5 min. Pelleted material was then resuspended in 18% (w/v) dextran solution and centrifuged at 4400 × g for 15 min. The pellet (brain vessel) was carefully separated from the supernatant (vessel-depleted brain). The pellet, containing the crude vascular fraction, was resuspended in Hank’s Balanced Salt Solution (HBSS). Vascular suspensions were next passed through a 100 μm nylon mesh filter to eliminate the larger, macrovascular components. The resulting filtrates were passed through a 40 μm cell strainer. The unfiltered microvessels were harvested by washing into a low binding tube and resuspended in HBSS. The brain microvessels were then resuspended in ice-cold lysis buffer, sonicated, centrifuged at 20,000 × g for 20 min, and supernatant was used for ASM activity and ceramide analysis.
ASM activity assays
We performed the enzymatic activity measurements as previously described17,21 using a UPLC system (Waters). Briefly, the brain was lysed in homogenization buffer containing 50 mM HEPES (Sigma-Aldrich, H3375), 150 mM NaCl (Sigma-Aldrich, S3014), 0.2% Igepal CA-630 (Sigma-Aldrich, I8896), and protease inhibitor (Calbiochem, 539131). Three microliters of the samples (plasma, serum, or brain) were mixed with 3 µl of 200 µM Bodipy-C12-sphingomyelin (Invitrogen, D7711) diluted in 0.2 M of sodium acetate buffer, pH 5.0, 0.2 mM ZnCl2, and 0.2% Igepal CA-630 and incubated at 37 °C for 1 h. The hydrolysis reactions were stopped by adding 114 µl of ethanol, and centrifuged at 15,493 × g for 5 min. Thirty microliters of the supernatant was then transferred to a sampling glass vial and 5 µl was applied onto a UPLC system for analysis. Quantification was achieved by comparison to Bodipy-C12-ceramide using the Waters Millennium software.
LC-MS/MS for ceramide quantification
We simultaneously analyzed Cer-16, Cer-18, Cer-20, Cer-22, and Cer-24 using a Agilent 6470 triple quadrupole liquid chromatography-mass spectrometry (LC–MS/MS) system (Agilent, Wilmington, DE, USA) by the modified method of previous study55. Briefly, standard calibration curves for five ceramides were prepared using the mixtures of Cer-16 (Avanti, 860516), Cer-18 (Avanti, 860518), Cer-20, (Avanti, 860520), Cer-22 (Avanti, 86051), and Cer-24 (Avanti, 860524) stock solution in the range of 0.5–1000 ng ml−1 of ceramides in HBSS. Standard curves for Cer-16, Cer-18, Cer-20, Cer-22, and Cer-24 showed good linearity (r2 > 0.996 for all ceramides) and the coefficient of variance for the interday precision and accuracy was below 15 %. For the ceramide analysis, aliquots (50 µl) of plasma and protein samples extracted from CD4+ T cell membrane were added to 200 μl of internal standard solution (IS; berberine 0.1 ng ml−1 in methanol) and vortexed for 5 min. After centrifugation of the mixture at 16,000 × g for 5 min, aliquots (5 μl) of the supernatant were injected into a LC–MS/MS system. Cer-16, Cer-18, Cer-20, Cer-22, and Cer-24 and berberine (IS) was separated on a Synergi Polar RP column (2.0 × 150 mm, 4 μm particle size; Phenomenex, Torrence, CA, USA) using a isocratic elution of distilled water containing 0.1% formic acid: methanol containing 0.1% formic acid = 10:90 (v/v) at a flow rate of 0.25 ml min−1. Quantification of the analyte peaks was carried out at m/z 538.5 → 264.3 for Cer-16 (retention time (TR) 4.6 min), m/z 566.5 → 264.3 for Cer-18 (TR 5.2 min), m/z 594.6 → 264.3 for Cer-20 (TR 6.3 min), m/z 622.6 → 264.3 for Cer-22 (TR 7.5 min), m/z 650.7 → 264.3 for Cer-24 (TR 9.2 min), and m/z 336.1 → 320.0 for berberine (IS, TR 2.2 min) in a positive ionization mode with optimized fragmentor of 120–135 V and collision energy of 25–35 eV, respectively. Concentrations of ceramides in the plasma and protein samples extracted from CD4+ T cell membrane were calculated from the linear regression equation of each ceramide standard curves using the ratio of the peak areas of ceramides and IS.
T cell differentiation
Naive CD4+ T cells were purified from spleen using CD4+ T cell isolation kit (Miltenyi biotec, 130-104-453) for in vitro and in vivo experiments. To extract protein of cell membrane and cytosol from isolated naive CD4+ T cells, mem-per plus membrane protein extraction kit (Thermo Fisher Scientific, 89848) was used. In some experiments, isolated naive CD4+ T cells were cultured in 6-well or 24-well plates (Costar) in RPMI 1640 medium (Gibco, 11875093) supplemented with 10% FBS (Gibco, 16000-044), 1% P/S (Gibco, 15140122) and 55 μM β–mercaptoethanol (21985-023), and stimulated with plate-bound anti anti-mouse CD3 (2 µg ml−1, Invitrogen, 16-0032-85) and anti-mouse CD28 (1 µg ml−1, Invitrogen, 16-0281-85). To investigate CD4+ T cell apoptosis by activated- or inactivated-ASM and ceramide, the serum (5%) from WT, Tie2-cre; Smpd1ox/ox, or Smpd1−/− mice were added to the naive CD4+ T cells. Apoptotic cells were detected flow cytometry using apoptosis kit (Invitrogen, V13242). To Th17 cell differentiation, naive CD4+ T cells were stimulated in the presence of IL-6 (20 ng ml−1), IL-23 (10 ng ml−1), IL-1β (10 ng ml−1), TGF-β1 (2 ng ml−1), anti-IL-4 (10 ng ml−1), anti-IFN-γ (10 µg ml−1), and anti-IL-2 (10 µg ml−1). To Treg cells differentiation, naive CD4+ T cells were stimulated in the presence of TGF-β1 (5 ng ml−1). All the cytokines are from Miltenyi biotec. The serum (5 %) from WT, Tie2-cre; Smpd1ox/ox, or Smpd1−/− mice were added to the cultures under the Th17 cell or Treg cell-polarizing condition. To confirm the effects of C16-ceramide, recombinant human ASM (rASM), or phosphorlycoline on Th17 cell differentiation, C16-creamide (Avanti, 860516), rASM (Genscript), or phosphorlycholine (MedChemExpress, HY-B2233B) was treated at 10 μM, 2.5 μM, and 10 μM under the Th17 cell-polarizing condition. To examine the inhibitory effects of plasma ASM by ASM antibody (23A12C3, generated for this study) on Th17 cell differentiation, IgG isotype control antibody (50 μg ml−1, R&D system, MAB002), ASM antibody (50 μg ml−1), and the serum (5%) of Tie2-cre; Smpd1ox/ox was treated to the cultures under the Th17 cell-polarizing condition. After 4 days, cells were collected for flow cytometry, real-time PCR, or western blot.
Co-cultures of Th17 cells and BV2 microglial cells
Th17 cells were differentiated with or without rASM (2.5 μΜ, Genscript) and IL17 antibody (30 μg ml−1, R&D system, MAB421) as described above. Before 1 day of co-culture with Th17 cells, BV2 microglial cells (Accegen, ABC-TC212S) were seeded in 6-well or 24-well plates (Costar) in RPMI 1640 medium (Gibco, 11875093) supplemented with 10% FBS (Gibco, 16000-044) and 1% P/S (Gibco, 15140122). BV2 microglial cells were co-cultured with Th17 cells for 1 day, and then BV2 microglial cells were collected for real-time PCR, morphology analysis, and Aβ phagocytosis assay. For in vitro Aβ phagocytosis assay, Hilyte Fluor 555-labeled-Aβ1-42 (AnaSpec, AS-60480) was aggregated for 24 h at 37 °C. Before 1 day of co-culture with Th17 cells, BV2 microglial cells were seeded in 24-well plates (Costar) in DMEM medium (Sigma-Aldrich, D5796) supplemented with 10% FBS and 1% P/S. BV2 microglial cells were co-cultured with Th17 cells for 1 day, and then Aβ (5 μg ml−1) was added and incubated for 8 h at 37 °C. For confocal measurement, anti-Iba1 antibody was used to label the cell shape and intracellular Aβ was quantified using MetaMorph software (Molecular Devices).
Flow cytometry
Immune cells in brain and blood were analyzed by flow cytometry. Single cells from brain were prepared as previously described with minor modifications56. Brain was dissected and immediately transferred in ice-cold HBSS (Gibco). After gentle mincing, the brain was digested in a HBSS solution containing collagenase P (0.2 mg ml−1, Roche), dispase II (0.8 mg ml−1, Roche), DNase I (0.01 mg ml−1, Roche), and collagenase A (0.3 mg ml−1, Roche) at 37 °C for 1 h under gentle rocking. Digestion was stopped by adding FBS (Gibco) on ice. The supernatants were centrifuged at 250 × g for 10 min at 4 °C. The pellet was resuspended in 25 % BSA (Gibco)/PBS (Gibco) for myelin removal. Following a centrifugation step at 3000 × g for 30 min at 4 °C, the myelin containing supernatant was discarded. The cell pellets were then resuspended in 1 ml of HBSS and filtered through a 40 μm mesh, followed by a washing step in HBSS. The cell pellets were resuspended in 1 ml of red blood cell lysis buffer (BD Biosciences) and incubated at RT for 10 min for lysis of erythrocytes. Subsequently, 2 ml of MACs buffer (Miltenyl Biotec, 130-091-222) was added and centrifuged at 250 × g for 10 min at 4 °C. Blood cells were prepared as previously described with minor modifications57. To obtain peripheral blood mononuclear cells (PBMCs), blood was collected in sodium-heparin tube (BD Biosciences, 367871) by cardiac puncture and blood was gently layered in the top of histopaque (Sigma-Aldrich, 10771). After centrifuge (400 × g, 30 min), PBMCs formed in the interphase between histopaque and plasma were collected and washed once with PBS. For analysis of myeloid immune cells, red blood cells were lysed once at 4 °C for 10 min in 0.15 M NH4Cl (STEMCELL Technologies) and washed once with PBS. The cells were stained with the following antibodies for blood and brain macrophage subsets56,58, monocytes58,59, neutrophils58,60, T cells61, Treg cells, and B cells62: mouse anti-CD11b APC (1:100, BD Bioscience, 553312), mouse anti-CD115 PE (1:100, Thermo Fisher Scientific, 12-1152-82), mouse anti-Ly6C FITC (1:100, BD Bioscience, 553104), and mouse anti-Ly6G APC-Cy7 (1:100, BD Bioscience, 557661), mouse anti-Ly6G FITC (1:100, BD Bioscience, 551460), mouse anti-lineage biotin (1:10, Miltenyl Biotec, 130-090-858), anti-biotin streptavidin PB (1:100, Invitrogen, S11222), mouse anti-CD11b PE (1:100, BD Bioscience, 557397), mouse anti-F4/80 APC (1:100, Thermo Fisher Scientific, 14-4801-82), mouse anti-CD45 PerCp Cy5.5 (1:100, BD Bioscience, 550994), mouse anti-CD11b PE (1:100, BD Bioscience, 557397), mouse anti-CD11b PeCy5 (1:100, Tonbo Bioscience, 55-011), mouse anti-CD45 APC-Cy7 (1:100, BD Bioscience, 557659), mouse anti-MHCII PE (1:100, BD Bioscience, 557000), mouse anti-CD206 PE-Cy7 (1:100, Thermo Fisher Scientific, 25-2061-82), mouse anti-CD4 FITC (1:100, BD Bioscience, 553047), mouse anti-CD8 APC (1:100, eBioscience, 17-0081-82), mouse anti-CD25 PE (1:100, eBioscience, 12-0251-82), mouse anti-FoxP3 APC (1:100, eBioscience, 17-5773-82), and mouse anti-B220 PE (1:100, Tonbo Bioscience, 50-0452). For staining of intracellular cytokines, single cells from brain or PBMCs was stimulated with RPMI/10% FBS/1% P/S/ with phorbol myristate acetate (PMA, 50 ng ml−1, Sigma-Aldrich, P8139), ionomycin (1 μg ml1, Sigma-Aldrich, I0634), Golgi stop (x1500, BD Bioscience, 554724), and Golgi plus (x1000, BD Bioscience, 555029), and incubated for 3 h. Cells were washed and stained with mouse anti-CD4 FITC (1:100, BD Bioscience, 553047) for 30 min. Permeabilization, fixation, and staining of intracellular cytokines with mouse anti-IFNγ APC (1:100, BD Bioscience, 553047), mouse anti-IL4 PE (1:100, Bio Legend, 504104), and mouse anti-IL17 PerCp Cy5.5 (1:100, BD Bioscience, 553142) were performed with Inside Stain Kit (Miltenyi Biotec, 130-090-477) according to the manufacturer’s instructions. Analysis was performed using Attune NxT flow cytometer (Thermo Fisher) and further analyzed using FlowJo analytical software (Tree Star, Inc.). Gating strategy for immune cells in blood, spleen, and brain was shown in Supplementary Figs. 14–16.
Histological analysis
For immunofluorescence staining, brain was cut on a vibratome (30 μm). Thioflavin S (Sigma-Aldrich, T1892) staining was carried out according to previously described procedures17,21. The following antibodies were used: 6E10 (mouse, 1:100, Signet, SIG39300), SMA (mouse, 1:400, Sigma-Aldrich, A2547), Iba1 (rabbit, 1:500, Wako, 019-19941), GFAP (rabbit, 1:500, Dako, N1506), Lamp1 (mouse, 1:200, Abcam, ab24170), and Fibrinogen (Fibrin, rabbit, 1:500, Dako, A008002). All were visualized using Alexa anti-mouse 488 and 633 or Alexa anti-rabbit 488 and 594 as secondary antibodies. To visualize brain microvessels, section was incubated with fluorescein labeled L. esculentum lectin (1:100, Vector Laboratories, FL-1171), CD31 (goat, 1:100, R&D system, AF3628), Collagen IV (rabbit, 1:100, Abcam, ab6586), and Aquaporin-4 (chicken, 1:100, Synaptic Systems, 429006). The sections were analyzed with a laser-scanning confocal microscope (FV3000; Olympus) or with a BX51 microscope (Olympus). For quantification of immunostaining of cortex and hippocampus, four images in each region were obtained as shown in Supplementary Fig. 17, and then intensity or cell count was quantified by using “Set color threshold tool” and “Show region statistics tool” of MetaMorph software (Molecular Devices). IMARIS software (Bitplane)21 was used for analysis of three-dimensional reconstruction of microglia. Confocal images were taken through a z-stack (total z-axis length = 10 μm) and were imported into the IMARIS software. Cell body width was measured, and cell dendrites were automatically detected using the analysis tool. Then, the image was converted into a 3D image, and the cell body volume, process length, number of branches, and terminal tips were automatically quantified. To quantification of amyloid angiopathy stained with Thioflavin S and SMA antibody, four images were taken at 40x magnification within cortex and hippocampus, and the percentage of Aβ intensity within each vessel stained with SMA antibody was measured by MetaMorph software.
Th17 cell transplantation
To confirm blood-derived Th17 cells in the brain, splenic naive CD4+ T cells, which are EGFP–, were purified from Il17a EGFP mice using a CD4+ T cell isolation kit (Miltenyi biotec, 130-104-453) and cultured in rASM (2.5 μM) under Th17 cell-polarizing conditions (IL-6 (20 ng ml−1), IL-23 (10 ng ml−1), IL-1β (10 ng ml−1), TGF-β1 (2 ng ml−1), anti-IL-4 (10 ng ml−1), anti-IFN-γ (10 µg ml−1), and anti-IL-2 (10 µg ml−1)) for 4 days to generate pathogenic EGFP+ Th17 cells. Three-month-old APP/PS1 mice were intraperitonially injected with IgG isotype control or IL17 antibody (50 μg per mouse, every other day) during 5 weeks of exposure to blood of ASM overexpressing Tie2-cre; Smpd1ox/ox mice. One week before sampling, cultured Th17 cells were intravenously injected (1 × 106 cells per mouse) into APP/PS1 mice in parabiosis with Tie2-cre; Smpd1ox/ox mice and treated with IgG isotype control or IL17 antibody. The brain of each mouse was cut on a vibratome (30 μm), and the sections were then stained with GFP (rat, 1:1000, Abcam, ab13970) and collagen IV (rabbit, 1:100, Abcam, ab6586) antibodies for quantification of perivascular and parenchymal EGFP+ Th17 cells. The sections were analyzed with a laser-scanning confocal microscope (FV3000; Olympus). Cell counts were obtained using MetaMorph software (Molecular Devices). Cell numbers per brain region were divided by the respective tissue area and represented as cells/mm2.
Th17 cell staining with BBB
FITC-labeled albumin (Sigma-Aldrich, A9771) was dissolved in buffered saline (10 mg/ml), and the fluorescent dye solution was slowly infused into the tail vein (10 ml/kg) as previously described63. With a three-minute interval after the infusion completion, the mice were killed by decapitation and their brains were fixed for 48 h in 4% paraformaldehyde in PBS. Brain was cut on a vibratome (30 μm). Leakage of FITC-labeled albumin into brain parenchyma was observed under a fluorescent microscope. These sections additionally immunoreacted with anti-RORγ antibody (1:100, Rabbit, Abcam, ab207082) determination of Th17 cells in the perivascular and parenchyma of cortex and hippocampus. Cell counts were obtained using MetaMorph software (Molecular Devices). Cell numbers per brain region were divided by the respective tissue area and represented as cells/mm2.
Western blotting
Samples were lysed in RIPA buffer (Cell signaling Technologies, 9806), then subjected to SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked with 5% milk, incubated with primary antibody and then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody17,21. Primary antibodies to the following proteins were used: APP (mouse, 1:500, Signet, SIG39300), BACE-1 (mouse, 1:1,000, Millipore, MAB5308), Synaptophysin (rabbit, 1:2000, Abcam, ab32127), PSD95 (mouse, 1:1000, Millipore, MAB1596), Synapsin 1 (rabbit, 1:1000, Synaptic systems, 106 103), MAP2 (chicken, 1:10000, Abcam, ab5392), p-Stat3 (rabbit, 1:1000, Cell Signaling Technology, 9145), Stat3 (mouse, 1:1000, Cell Signaling Technology, 9139), p-JNK (mouse, 1:1000, Cell Signaling Technology, 9255), JNK (rabbit, 1:1000, Cell Signaling Technology, 9252), p-Akt (rabbit, 1:1000, Cell Signaling Technologies, 4060), Akt (rabbit, 1:1000, Cell Signaling Technologies, 4091), p-mTOR (rabbit, 1:1000, Cell Signaling Technology, 5536), mTOR (rabbit, 1:1000, Cell Signaling Technology, 2983), Fibrinogen (Fibrin, rabbit, 1:500, Dako, A008002), Thrombin (goat, 1:100, Santa Cruz Biotechnology, sc-23355), ZO-1 (rabbit, 1:500, Invitrogen, 40-2200), Occludin (mouse, 1:500, Invitrogen, 33-1500), Claudin5 (mouse, 1:500, Invitrogen, 35-2500), and β-actin (1:1,000, Santa Cruz, SC-1615). Rabbit-HRP (1:1000, Cell signaling, 7074), goat-HRP (1:1000, Santa Cruz Biotechnology, sc2020) and mouse-HRP (1:1000, Cell signaling, 7076) were used as secondary antibody. We performed densitometric quantification using the ImageJ software (National Institutes of Health). Images have been cropped for presentation.
ELISA
For measurement of Aβ40, Aβ42, and IL17, we used commercially available ELISA kits (Invitrogen, KHB3481 for Aβ40; Invitrogen, KHB3441 for Aβ42; and R&D system, M1700 for IL17). Cortex and hippocampus of mice were homogenized in buffer containing 0.02 M guanidine. ELISA was then performed for Aβ40, Aβ42 and IL17 according to the manufacturer’s instructions.
Immunization with ASM peptide and administration of ASM antibody
To examine the possible prophylactic effects of plasma ASM-targeting active immunotherapy, 50 μg/100 μl of ASM protein or 100 μl PBS was mixed with 100 μl of the complete freund’s adjuvant (Sigma-Aldrich, F5881) and repeatedly passed through a micro-emulsifying needle (Cadence Science, CAD7977) until the mixture became pasty. Then, 200 μl of mixture was injected intraperitoneally (i.p.) into 6-mo-old WT and APP/PS1 mice. Second and third immunization was performed every 2 weeks with 25 μg/50 μl of ASM protein or 50 μl PBS mixed with 50 μl of the incomplete freund’s adjuvant (Sigma-Aldrich, F5506). Four weeks after third immunization, mice were immunized 25 μg/50 μl of ASM protein or 50 μl PBS mixed with 50 μl of the incomplete freund’s adjuvant. After 4 weeks, mice were sacrificed for analysis. For passive immunotherapy, IgG isotype control antibody (50 mg kg−1, R&D system, MAB002) or ASM antibody (50 mg kg−1, 23A12C3, generated for this study) was injected twice a week i.p. for 8 weeks to 7-mo-old APP/PS1 mice until the age of 9 months.
In vivo multiphoton microscopy
In vivo multiphoton experiments were performed as previously described17. After anesthesia, a cranial window was placed over the partial cortex. Blood plasma was labeled by tail vein injection of TMR-dextran (MW = 40 kD; Invitrogen, D1842). In vivo time-lapse images were acquired at 2, 15, and 30 min after TMR-dextran injection. The leakage from cortical vessels (layer II and III, approximately 100 μm from the cortical surface) was captured in each mouse. Quantification was performed by a blinded investigator by measuring the fluorescent signal intensity in 20 randomly selected 20 μm × 20 μm extravascular areas in brain parenchyma using the NIH ImageJ software integrated density function.
Behavioral studies
We performed behavioral studies to assess spatial learning and memory in the Morris water maze as previously described17,21. Animals were given four trials per day for 10 d to learn the task. At day 11, animals were given a probe trial in which the platform was removed. Fear conditioning was conducted by previously described techniques17,21. On the conditioning day, mice were individually placed into the conditioning chamber. After a 60 s exploratory period, a tone (10 kHz, 70 dB) was delivered for 10 s; this served as the conditioned stimulus (CS). The CS co-terminated with the unconditioned stimulus (US), a scrambled electrical footshock (0.3 mA, 1 s). The CS-US pairing was delivered twice at a 20 s intertrial interval. On day 2, each mouse was placed in the fear-conditioning chamber containing the same exact context, but with no administration of a CS or footshock. Freezing was analyzed for 5 min. On day 3, a mouse was placed in a test chamber that was different from the conditioning chamber. After a 60 s exploratory period, the tone was presented for 60 s without the footshock. The rate of freezing response of mice was used to measure the fear memory. All results of behavioral experiments were collected from male mice.
Recombinant human ASM and monoclonal ASM antibody generation
The synthesis of recombinant human ASM (rASM) and production of mouse monoclonal ASM antibodies were performed by Genscript. For synthesis of rASM, Expi293F cells (Thermo Fisher Scientific, A14527) were transfected with the full-length human ASM cDNA plasmid. The cell culture supernatants were collected on day 6 were used for purification. Cell culture broth was centrifuged. Cell culture supernatant was loaded onto an affinity purification column at an appropriate flow rate. After washing and elution with appropriate buffers, the eluted fractions were pooled and buffer exchanged to the final formulation buffer. The purified protein was analyzed by SDS-PAGE, Western blot analysis to determine the molecular weight (68 kDa) and purity. For production of mouse monoclonal ASM antibodies, screening of 110 hybridomas was performed by ELISA against rASM. Positive clones were expanded and re-tested to confirm epitope reactivity to rASM. Antibody clones were screened through IC50 determinations as described below.
IC50 determinations
The fluorescent ASM assay was performed in a 96 well plate using HNPPC (2-N-Hexadecanoyl-4-nitrophenylphosphorylcholine, Toronto Research Chemicals, 60438-73-5) as the substrate. ASM and HNPPC were diluted to 2 µg ml−1 and 1 mM in assay buffer (50 mM MES, 0.5 µM ZnCl2, pH 6.5) and incubated for 10 min at 37 °C. ASM antibody (23A12C23) with various concentrations (1 nM to 500 nM) were pre-incubated for 60 min at 37 °C together with 2 µg ml−1 ASM, and then HNPPC was added. For the standard curve, p-nitrophenol was used (Sigma-Aldrich, 241326). After incubation for 6 h at r.t., the reaction was stopped by addition of develo** buffer (0.2 M NaOH) and the absorbance of HNPPC was measured at 410 nm. The ASM activity was then calculated: [substrate blank (OD) × conversion factor (pmol/OD, derived using calibration standard p-nitrophenol)/incubation time (h) × amount of enzyme (µg)]. The IC50 was analyzed using the GraphPad Prism 7.0 software. Each experiment was performed in triplicate.
Monoclonal ASM antibody binding assay and ASM titer assay
The recombinant human ASM (2 µg ml−1, Genscript) in ELISA coating buffer (Abcam, ab210899) was coated onto MaxiSorp ELISA plates (Thermo Fisher Scientific) overnight at 4 °C. Wells were incubated with blocking buffer (Abcam, ab210904) for 2 h at RT, washed with wash buffer (Abcam, ab206977). ASM antibody (23A12C23) or diluted plasma in blocking buffer were incubated for 2 h at RT. Following washing with wash buffer, Anti-mouse IgG/Biotin (Sigma-Aldrich, B7264) in blocking buffer was added and incubated for 1 h at RT. After standard washing, wells were further incubated with streptavidin HRP (Abcam, ab210901) in blocking buffer for 1 h at RT and results were developed with TMB substrate (Abcam, ab210902). Absorbance was measured at 450 nm using a Varioskan LUX Multimode Microplate Reader (Thermo Scientific). The KD was analyzed using the SigmaPlot 13.0 software. Each experiment was performed in triplicate. The ASM titers were defined as the dilution factor referring to 50 % of the maximal optical density (ODmax/2).
Surface plasmon resonance (SPR) spectroscopy
SPR binding experiments were performed using a Biacore® T200 instrument (Biacore, now GE Healthcare). Recombinant ASM was kindly provided by Prof. Edward H. Schuchman (Icahn School of Medicine at Mount Sinai, New York, New York, USA)64. ASM was immobilized on the surface of a CM5 sensor chip (GE Healthcare) utilizing standard amine coupling chemistry. The CM5 sensor chip surface was activated by an injection of 0.4 M EDC and 0.1 M NHS at 10 µl min−1 for 420 s. HBS-EP buffer containing was used as the running buffer with pH 7.4 (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, and 0.005% v/v surfactant P20). ASM (theoretical pI = 6.8) at 25 µg ml−1 in 10 mm sodium acetate, pH 4.5, injected over the activated surface at 10 µl min−1 for 600 s. The amount of ASM immobilized on the activated surface was typically 5500 response units (RU). The excess hydroxysuccinimidyl groups on the surface were deactivated with 1 M ethanolamine hydrochloride, pH 8.5 for 420 s at a flow rate of 10 µl min−1. The surface of a reference flow cell was activated with 0.4 M EDC/0.1 M NHS for 420 s with a flow rate of 10 µl min−1, and then deactivated with a 420 s exposure of 1 M ethanolamine at a flow rate of 10 µl min−1. With no ligand bound to the flow path, the control flow cell was used to detect nonspecific binding of the small molecules to the sensor chip surface during screening affinity assays. The ASM antibody (23A12C23) were diluted with the assay buffer (10 mM phosphate buffer, 137 mM NaCl, 2.7 mM KCl, pH 7.4, 0.05% v/v surfactant P20) to yield antibody solutions for the assay of concentrations that varied from 0.195 nM to 12.5 nM. Prior to analyte injection, the series S CM5 chip was conditioned with three 30 s cycles of assay buffer followed by three startup cycles, allowing the response to stabilize before analyte injection. Data were collected at a temperature of 25 °C and individual antibody samples were tested from lowest to highest concentrations. During each sample cycle, analyte was injected for 150 s at a flow rate of 10 µl min−1. A dissociation period was monitored for 300 s after analyte injection to wash any remaining analyte from the sensor chip before running the next sample. The Biacore T200 was programmed to run an automated assay with the various antibody samples. The responses measured in the blank flow cell (control) were subtracted from the response measured in the flow cell with protein immobilized. Equilibrium constants (KD) were calculated using the ‘kinetic’ model in Biacore T200 Evaluation Software. All experiments were repeated three times.
RNA isolation and real-time PCR analysis
RNA was extracted from the brain homogenates and cell lysates using the RNeasy Lipid Tissue Mini kit and RNeasy Plus Mini kit (QIAGEN) according to the manufacturer’s instructions. cDNA was synthesized from 5 μg of total RNA using a commercially available kit (Takara Bio Inc.). Quantitative real-time PCR was performed using a Corbett research RG-6000 real-time PCR instrument. Used primers are described in Supplementary Table 2.
Statistical analysis
Sample sizes were determined by G-Power software (ver 3.1.9.4, with α = 0.05 and power of 0.8). In general, statistical methods were not used to re-calculate or predetermine sample sizes. Variance was similar within comparable experimental groups. Individuals performing the experiments were blinded to the identity of experimental groups until the end of data collection and analysis for at least one of the independent experiments. All data are representative of at least three independent experiments. Comparisons between two groups were performed with a two-tailed student’s t test. In cases where more than two groups were compared to each other, one way analysis of variance (ANOVA) was used, followed by Tukey’s HSD test. All statistical analyses were performed using GraphPad Prism 7.0 software. P < 0.05 were considered as statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the article and its Supplementary Information. The datasets generated and/or analyzed during the current study are also available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186 (2016).
Hu, W. T. et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 79, 897–905 (2012).
Kang, S., Moser, V. A., Svendsen, C. N. & Goodridge, H. Rejuvenating the blood and bone marrow to slow aging-associated cognitive decline and Alzheimer’s disease. Commun. Biol. 3, 69 (2020).
Ma, J. et al. Circulating factors in young blood as potential therapeutic agents for age-related neurodegenerative and neurovascular diseases. Brain Res. Bull. 153, 15–23 (2019).
Hsiao, Y. T., Shimizu, I., Yoshida, Y. & Minamino, T. Role of circulating molecules in age-related cardiovascular and metabolic disorders. Inflamm. Regen. 42, 2 (2022).
Hye, A. et al. Plasma proteins predict conversion to dementia from prodromal disease. Alzheimers Dement. 10, 799–807 (2014).
Villeda, S. A. et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663 (2014).
Katsimpardi, L. et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634 (2014).
**a, E. et al. Young blood rescues the cognition of Alzheimer’s model mice by restoring the hippocampal cholinergic circuit. Neuroscience 417, 57–69 (2019).
Middeldorp, J. et al. Preclinical assessment of young blood plasma for Alzheimer disease. JAMA Neurol. 73, 1325–1333 (2016).
Sha, S. J. et al. Safety, tolerability, and feasibility of young plasma infusion in the plasma for Alzheimer symptom amelioration study: a randomized clinical trial. JAMA Neurol. 76, 35–40 (2019).
Villeda, S. A. et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011).
Smith, L. K. et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 21, 932–937 (2015).
Yousef, H. et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 25, 988–1000 (2019).
Bu, X. L. et al. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 23, 1948–1956 (2018).
Morales, R. et al. Infusion of blood from mice displaying cerebral amyloidosis accelerates amyloid pathology in animal models of Alzheimer’s disease. Acta Neuropathol. Commun. 8, 213 (2020).
Park, M. H. et al. Vascular and Neurogenic Rejuvenation in Aging Mice by Modulation of ASM. Neuron 100, 167–182 (2018).
Lee, J. K. et al. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 211, 1551–1570 (2014).
Kornhuber, J., Rhein, C., Müller, C. P. & Mühle, C. Secretory sphingomyelinase in health and disease. Biol. Chem. 396, 707–736 (2015).
Ong, W. Y., Herr, D. R., Farooqui, T., Ling, E. A. & Farooqui, A. A. Role of sphingomyelinases in neurological disorders. Expert. Opin. Ther. Targets 19, 1725–1742 (2015).
Park, M. H. et al. Discovery of a dual-action small molecule that improves neuropathological features of Alzheimer’s disease mice. Proc. Natl Acad. Sci. U.S.A. 119, e2115082119 (2022).
Gulbins, E. et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat. Med. 19, 934–938 (2013).
Jenkins, R. W., Canals, D. & Hannun, Y. A. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 21, 836–846 (2009).
Park, M. H., **, H. K. & Bae, J. S. Potential therapeutic target for aging and age-related neurodegenerative diseases: the role of acid sphingomyelinase. Exp. Mol. Med. 52, 380–389 (2020).
Howlett, D. R. et al. Cognitive correlates of Abeta deposition in male and female mice bearing amyloid precursor protein andpresenilin-1 mutant transgenes. Brain Res. 1017, 130–136 (2004).
Wyss-Coray, T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015 (2006).
Schwartz, M., Kipnis, J., Rivest, S. & Prat, A. How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci. 33, 17587–17596 (2013).
Baik, S. H. et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 35, 1286–1292 (2014).
Michaud, J. P., Bellavance, M. A., Préfontaine, P. & Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 5, 646–653 (2013).
Tahmasebinia, F. & Pourgholaminejad, A. The role of Th17 cells in auto-inflammatory neurological disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 79, 408–419 (2017).
Zhu, J. & Paul, W. E. CD4 T cells: fates, functions, and faults. Blood 112, 1557–1569 (2008).
Luckheeram, R. V., Zhou, R., Verma, A. D. & **a, B. CD4+ T cells: differentiation and functions. Clin. Dev. Immunol. 2012, 925135 (2012).
Horinouchi, K. et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat. Genet. 10, 288–293 (1995).
Korn, T., Bettelli, E., Oukka, M. & Kuchroo, V. K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517 (2009).
Zhou, L. & Littman, D. R. Transcriptional regulatory networks in Th17 cell differentiation. Curr. Opin. Immunol. 21, 146–152 (2009).
Ghoreschi, K. et al. Generation of pathogenic TH17 cells in the absence of TGF-b signalling. Nature 467, 967–971 (2010).
Codarri, L. et al. RORct drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 12, 560–567 (2011).
Reboldi, A. et al. C–C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 10, 514–523 (2009).
Bai, A. & Guo, Y. Acid sphingomyelinase mediates human CD4+ T-cell signaling: potential roles in T-cell responses and diseases. Cell Death Dis. 8, e2963 (2017).
Miossec, P. & Kolls, J. K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 11, 763–776 (2012).
Maddur, M. S., Miossec, P., Kaveri, S. V. & Bayry, J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am. J. Pathol. 181, 8–18 (2012).
Kebir, H. et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 10, 1173–1175 (2007).
Rahman, M. T. et al. IFN-g, IL-17A, or zonulin rapidly increase the permeability of the blood-brain and small intestinal epithelial barriers: Relevance for neuro-inflammatory diseases. Biochem. Biophys. Res. Commun. 507, 274–279 (2018).
Adam, D., Heinrich, M., Kabelitz, D. & Schütze, S. Ceramide: does it matter for T cells? Trends Immunol. 23, 1–4 (2002).
Cipollini, V., Anrather, J., Orzi, F. & Iadecola, C. Th17 and Cognitive Impairment: Possible Mechanisms of Action. Front Neuroanat. 13, 95 (2019).
Solleiro-Villavicencio, H. & Rivas-Arancibia, S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+ T cells in neurodegenerative diseases. Front. Cell. Neurosci. 12, 114 (2018).
Cristiano, C. et al. Neutralization of interleukin-17 rescues amyloid-b-induced neuroinflammation and memory impairment. Br. J. Pharmacol. 176, 3544–3557 (2019).
Oberstein, T. J. et al. Imbalance of circulating Th17 and regulatory T cells in Alzheimer’s disease: a case control study. Front. Immunol. 9, 1213 (2018).
Tahmasebinia, F. & Pourgholaminejad, A. The role of Th17 cells in autoinflammatory neurological disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 79, 408–416 (2017).
Sevigny, J. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56 (2016).
Kondo, A. et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436 (2015).
Rajamohamedsait, H., Rasool, S., Rajamohamedsait, W., Lin, Y. & Sigurdsson, E. M. Prophylactic Active Tau Immunization Leads to Sustained Reduction in Both Tau and Amyloid-β Pathologies in 3xTg Mice. Sci. Rep. 7, 17034 (2017).
Atwal, J. K. et al. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci. Transl. Med. 3, 84ra43 (2011).
Yu, Y. J. & Watts, R. J. Develo** therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 10, 459–472 (2013).
Liebisch, G. et al. Quantitative measurement of different ceramide species from crude cellular extracts by electrospray ionization tandem mass spectrometry (ESI-MS/MS). J. lipid Res. 40, 1539–1546 (1999).
Scholz, A. et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 8, 39–57 (2016).
Park, M. H. et al. Neuropeptide Y Induces Hematopoietic Stem/Progenitor Cell Mobilization by Regulating Matrix Metalloproteinase-9 Activity Through Y1 Receptor in Osteoblasts. Stem Cells 34, 2145–2156 (2016).
Aryal, B. et al. ANGPTL4 deficiency in haematopoietic cells promotes monocyte expansion and atherosclerosis progression. Nat. Commun. 7, 12313 (2016).
Möhle, L. et al. Ly6C(hi) Monocytes Provide a Link between Antibiotic-Induced Changes in Gut Microbiota and Adult Hippocampal Neurogenesis. Cell Rep. 15, 1945–1956 (2016).
Zenaro, E. et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886 (2015).
Yang, K., Neale, G., Green, D. R., He, W. & Chi, H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol. 12, 888–897 (2011).
Knier, B. et al. Myeloid-derived suppressor cells control B cell accumulation in the central nervous system during autoimmunity. Nat. Immunol. 19, 1341–1351 (2018).
Zhang, J., Ke, K. F., Liu, Z., Qiu, Y. H. & Peng, Y. P. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of aβ1-42-induced Alzheimer’s disease model rats. PLoS ONE 8, e75786 (2013).
He, X. et al. Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim. Biophys. Acta 1432, 251–264 (1999).
Acknowledgements
We particularly would like to thank Yu Sin Han, Eun Yeong Lim, and Yun Ju Park for technical assistance. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2018M3C7A1056513 to H.K.J, 2020R1A2C3006875 to J.S.B, 2020R1A2C3006734 to H.K.J, 2020R1A4A2002691 to H.K.J). This research was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare and MSIT, Republic of Korea (HU20C0345 to J.S.B).
Author information
Authors and Affiliations
Contributions
B.J.C. and M.H.P. designed and performed experiments and wrote the paper. K.H.P., W.H.H., H.J.Y., H.Y.J., J.Y.H., M.R.C., and K.Y.K. performed experiments and analyzed data. J.L., I.S.S., M.P., and M.K.C. performed LC-MS/MS experiments and analyzed data. E.G., M.R. and J.K. generated and provided Smpd1 ox/ox mice. S.H.K. performed normal and AD patient plasma experiment. E.H.S. provided the Smpd1−/− mouse and recombinant ASM. C.W.H., C.K., S.H.K., E.H.S., H.K.J., and J.S.B. interpreted the data and reviewed the paper. H.K.J. and J.S.B. designed the study and wrote the paper. All authors discussed results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, B.J., Park, M.H., Park, K.H. et al. Immunotherapy targeting plasma ASM is protective in a mouse model of Alzheimer’s disease. Nat Commun 14, 1631 (2023). https://doi.org/10.1038/s41467-023-37316-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37316-z
- Springer Nature Limited
This article is cited by
-
Acid sphingomyelinase as a pathological and therapeutic target in neurological disorders: focus on Alzheimer’s disease
Experimental & Molecular Medicine (2024)
-
Inhibition of acid sphingomyelinase reduces reactive astrocyte secretion of mitotoxic extracellular vesicles and improves Alzheimer’s disease pathology in the 5xFAD mouse
Acta Neuropathologica Communications (2023)