

Abstract
Vascular endothelial cells (ECs) play a central role in the pathophysiology of many diseases. The use of targeted nanoparticles (NPs) to deliver therapeutics to ECs could dramatically improve efficacy by providing elevated and sustained intracellular drug levels. However, achieving sufficient levels of NP targeting in human settings remains elusive. Here, we overcome this barrier by engineering a monobody adapter that presents antibodies on the NP surface in a manner that fully preserves their antigen-binding function. This system improves targeting efficacy in cultured ECs under flow by >1000-fold over conventional antibody immobilization using amine coupling and enables robust delivery of NPs to the ECs of human kidneys undergoing ex vivo perfusion, a clinical setting used for organ transplant. Our monobody adapter also enables a simple plug-and-play capacity that facilitates the evaluation of a diverse array of targeted NPs. This technology has the potential to simplify and possibly accelerate both the development and clinical translation of EC-targeted nanomedicines.
Similar content being viewed by others


Introduction
Endothelial cells (ECs) are attractive therapeutic targets because they play an active role in a diverse array of diseases and are directly accessible to intravascular treatments1,2. Despite this potential, there are currently no EC-specific therapies in clinical use3, a fact driven by the challenge of treating ECs without affecting other cell types. Endothelial-targeted nanoparticles (NPs) are a promising solution, as they have the potential to deliver a concentrated dose of various therapeutics (e.g. small molecule, nucleic acid, protein) directly to targeted ECs. In addition, NPs can protect the encapsulated therapeutic from degradation by the environment, facilitate its transport across ECs membranes and sustain its release once inside the cells to prolong the duration of treatment4. Thus, effective targeting of NPs to the vascular endothelium within an organ of interest would create many new treatment possibilities across a variety of diseases.
The first hurdle to achieving EC-specific therapies is to localize the NPs to the tissue to be treated while avoiding losing NPs to the phagocytes of the liver and spleen5,6. In the field of organ transplant, we can circumvent this challenge by delivery in isolated organs during ex vivo normothermic machine perfusion (EVNMP). EVNMP is already in clinical use as a method to assess, preserve and potentially revive marginal organs with the goal of expanding access to organ transplantation7. The risk of dysfunctional inflammation is greater in marginal organs—organs from older and less healthy donors—and contributes to these organs being declined for transplant more frequently due to higher risk of post-transplant complications. Following organ recovery and preservation, ECs play a critical role in post-transplant pathologies associated with the dysfunctional inflammation. A single dose of therapeutics delivered in the forms of vascular-targeted NPs during EVNMP has the potential to reduce the immunogenicity of the graft and to provide several weeks of protection against dysfunctional inflammation during the post-transplant period when the organ is in its most vulnerable state8. Reduced endothelial activation has been demonstrated by treatment with anti-inflammatory molecules that inhibit NF-κB9,10,11, mTOR12, and complement13,14, PACE Poly(amine-co-ester), PVA Poly(vinyl alcohol), VS vinyl sulfone.
In the work shown here, we have utilized FCM101 which is specific for the mouse IgG1 isotype. Mouse IgG1 antibodies against human antigens are readily available, making this an ideal design to develop and demonstrate the approach. To minimize immune-reactions in humans, we envision the use of human or humanized Abs and a Mb adapter specific to human IgG Fc. Just as human Abs can be developed using many modern technologies, Mbs to human Fc can be developed following the well-established pipeline26,28. Furthermore, we envision that the potential problem of Ab displacement with endogenous Abs can be eliminated with a monobody adapter specific to a mutant Fc of human IgG (e.g. the so-called LALA variant that is commonly used for therapeutic antibodies42). We expect Mb adapters to minimally contribute to the overall immunogenicity of NPs based on clinical trials with a related molecule (PEGylated Adnectin) (NCT02515669, NCT03984812)43,44.
Ab-Mb-NPs also resulted in excellent vascular area coverage when perfused in human kidneys during EVNMP. This result is encouraging for future applications of therapeutic delivery to marginal organs, such as the kidneys enrolled in this study. Targeted NPs to treat marginal organs prior to transplant could reduce the risk of dysfunctional endothelial cell inflammation and associated recruitment of immune cells, rendering those organs safer to transplant and thereby increasing the number of transplantable organs. Management of peri-operative graft injuries, e.g., due to ischemia/reperfusion or to binding of graft-reactive alloantibodies in pre-sensitized recipients, with therapeutic NPs likely will require that a critical number of ECs are exposed to enough NPs to reach an effective therapeutic dose. In the context of EVNMP, this is only possible if NP binding efficiency is strong in complex 3D environments involving flow. In fact, develo** NPs in this setting will not only enable clinical translation in the context of ex vivo repair of marginal organs for transplant, but can also provide new insights relevant to intra-arterial or systemic delivery in the context of native kidney disease. Recent advances in EVNMP make it possible to sustain human organs outside the body for as long as 1 week45, which may allow us to evaluate safety and efficacy of vascular-targeted nanomedicines in a human context prior to exposing patients to risk in human clinical trials.
Ab-Mb-NPs have shown strong cellular binding and possess modularity to support rapidly develo** applications and their progression through experimental platforms. The Ab-Mb-NPs have the potential for almost immediate application in organ transplantation, and we are optimistic that this technology can be further developed for systemic delivery in a wide array of disease indications.
Methods
Chemicals
Poly (lactic acid)-poly (ethylene glycol) (PLA-PEG) block copolymer (Mw 16:5 kDa) were purchased from PolySciTech with different end group on the PEG block: carboxylic acid (PLA-PEG-COOH, AI030), methoxy (PLA-PEG-m, AK054) or maleimide (PLA-PEG-mal, AI065). Divinyl Sulfone (DVS, at 1.177 g/mL, <500 ppm hydroquinone as inhibitor), poly(vinyl alcohol) polymer (PVA 31–50 kDa, 87–89% hydrolyzed), sodium hydroxide (NaOH, reagent grade >98% anhydrous), chloridric acid (HCl, 37%), pentadecanolide (98%), N-Methyldiethanolamine (>99%), diethyl sebacate (98%), lipase acrylic resin from Candida antartica (>5,000 U/g, recombinant, expressed in Aspergillus niger), gold nanoparticles maleimide functionalized (30 nm) and dexamethasone were acquired by Sigma-Aldrich. Deuterium oxide (D2O, for NMR, 99,8 atom%D), phenyl ether (99%) were obtained from ACROS organic. 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), Hoechst 33258, DL-dithiolthreitol (DTT), LDS Sample buffer (4x), NuPAGETM SDS Running Buffer (20×), Fisherbrand superfrost microscope slides, cover slips (22 × 40 mm) were purchased from Thermo-Fisher Scientific. 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) was obtained from Botium. Dulbecco’s phosphate buffered saline (DPBS), medium 199 (M199), fetal bovine serum (FBS), penicillin-streptomycin, L-glutamine and TryPLE were acquired from Gibco. Coomassie Brilliant Blue R250 was obtained from OmniPur. Purified anti-human-CD31 monoclonal antibody (PECAM-1, IgG1 mouse, clone WM59), purified mouse IgG1 K isotype control antibody (clone MOPC-21) were purchased from Biolegend. Purified and FITC labeled anti-porcin-CD31 monoclonal antibody (PECAM-1, IgG1 mouse, clone LCI-4), purified anti-human-CD102 monoclonal antibody (ICAM-2, IgG1 mouse, clone BT-1), Alexa Fluor 488 labeled anti-human-CD105 monoclonal antibody (endoglin, IgG1 mouse, clone SN6) FITC labeled anti-human-CD31 monoclonal antibody (PECAM-1, IgG1 mouse, clone WM59), Alexa Fluor 647 labeled rabbit anti-Mouse IgG (H + L) cross-adsorbed secondary antibody were acquired from Invitrogen. BUV395 labeled anti-human-CD105 monoclonal antibody (endoglin, IgG1 mouse, clone 266) was obtained from BD Bioscience. Acetic acid glacial was purchase from RICCA. Methanol and dimethyl sulfoxide (DMSO) were acquired from JT Baker. Ethanol was obtained from AmericanBio. Human Umbilical Vein Endothelial Cells (HUVECs) were obtained from the Yale Vascular Biology and Transplantation tissue culture core laboratory, where they were isolated from fresh umbilical veins by treatment with collagenase. Porcine Aortic Endothelial Cells (PAECs) were purchased from Cell Applications, Inc. Endothelial Growth Supplement (ECGS) was acquired from Corning. Porcine EC Basal Medium with Porcine EC Growth Supplement was obtained from Cell Applications. Collagenase II was purchased from Worthington Biochemical. Human fibronectin was acquired from R&D. DyLight 649 labeled Ulex europaeus type I lectin (UEA I), Fluorescein labeled UEA1, antifade mounting medium with 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) were obtained from Vector Laboratories. Ringer’s lactate (LR) solution and sodium bicarbonate (8.4%) were purchased from Hospira. Mannitol (10%), Glucose (5%) and Ringer’s solution were acquired from Baxter Healthcare. Heparin (1000 IU/ml) was obtained from McKesson Medical-Surgical.
Monobody development
Mouse IgG1 Fc protein with C-terminal Avi-tag and His6 tag was produced in ExpiCHO cells (ThermoFisher) and purified using Ni-affinity chromatography. The mouse Fc protein was enzymatically biotinylated using purified BirA enzyme. This protein was used to screen monobody phage-display libraries28,46. After four rounds of phage selection, the sorted pools were subcloned into a yeast display vector after recombination of 5′ and 3′ fragments, followed by two rounds of sorting in the yeast display format28. The monobody clone, FCM101, was validated for target binding using yeast display28,46. FCM101 with an N-terminal tag containing His6, FLAG epitope, a TEV cleavage site and a C-terminal tag containing a cystein residue were expressed using the pHFT vector in Escherichia coli and purified using nickel-affinity chromatography28,46. The amino acid sequence of FCM101 is shown below with the N- and C-terminal tags underlined.
MKHHHHHHSSDYKDDDDKGENLYFQGSVSSVPTKLEVVAATPTSLLISWDAPAVTVYYYVITYGETGGNSPVQEFTVPGSKSTATISGLKPGVDYTITVYAGYGSGGYYSPISINYRTEIDKC. The expression vector for FCM101 has been deposited to Addgene.
PACE production
Poly(amine-co-ester) (PACE) was synthesized following previously published protocols by Kauffman and Piotrowski-Daspit36. Briefly, monomers (1.180 g MDEA, 2.558 g DES and 3.570 g PDL) together with enzyme (0.731 g lipase) were massed to obtain a molar ratio of 3:2:1. These materials were combined in a round bottom flask together with 14.6 g phenyl ether. Oligomerization was carried out under argon for 20 h. Polymerization was then carried out under vacuum for 48 h. The polymer was purified with three hexane washes to remove phenyl ether, and dissolved in DCM to remove the enzyme beads. Finally, DCM was evaporated under vacuum pressure using a rotary evaporator. The final product was characterized using NMR.
PVA-VS synthesis
PVA-VS polymer was prepared as described by Raudszus et al.47. The excess of divinyl sulfone was removed by dialysis for 24 h against water (Slide-A-Lyzer®Dialysis Cassette, 10 000 MCWO, Thermo Scientific). The resulting PVA-VS was lyophilized and characterized by 1H NMR in D2O (Agilent DD2 400 MHz).
Nanoparticles preparation and characterization
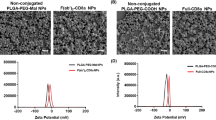
PLA-PEG NPs were prepared following a nanoprecipitation method17. PLA-PEG-COOH NPs were prepared with PLA-PEG carboxylic acid terminated polymer and PLA-PEG-mal NPs were prepared with a blend of methoxy and malemide terminated PLA-PEG polymer at a 1:1 weight ratio. Briefly, the polymer was dissolved in DMSO at 50 mg/mL and mixed with lipophilic fluorescent dye (DiI) at a dye:polymer weight ratio of 0.5%. This organic phase was then added dropwise with a glass pasteur pipette into distilled water under vigorous agitation (700 rpm, organic:aqueous phase volume ratio of 1:4). The organic solvent was subsequently removed using a 50 mL amicon filter tube (Amicon® Ultra-15 Centrifugal Filter Unit, MCWO 10000, Sigma Aldrich) for small batches or using a tangential flow filtration system (Spectrum® KrosFlo® KR2i TFF System, Repligen) equipped with a Microkros column (Microkros 20 cm 500 K MPES 0.5 mm MLL X FLL, C02-E500-05-N, Repligen) for big batches (more than 50 mg). The NPs suspension in DI water was flash frozen in liquid nitrogen and stored at −80 °C before use.
PACE-PVA-VS NPs were prepared following an emulsion-evaporation method36. The polymer was dissolved in DCM at 50 mg/mL and mixed with lipophilic fluorescent dye (DiD) at a dye:polymer weight ratio of 0.5%. This organic phase was then added dropwise with a glass pasteur pipette into a PVA-VS solution at 5% (w/w) under vigorous vortexing (organic:aqueous phase volume ratio of 1:2). The resulting emulsion was then transferred into a beaker containing 3 times its volume of PVA solution at 0.3% (w/w) under agitation. After 1 min the organic solvent was evaporated using a rotary evaporator. The excess of PVA and PVA-VS was washed by two centrifugations (18,000 × g, 45 min, 4 °C). The pellet was redispersed in DI water, flash frozen in liquid nitrogen and stored at −80 °C.
The size and potential zeta of the NPs were measured by dynamic light scattering (Zetasizer Nano ZS, He-Ne laser 633 nm, 173°, Smoluchowski equation, Malvern Panalytical).
Ab conjugation to nanoparticles using EDC-NHS chemistry
PLA-PEG-COOH NPs were conjugated to targeting antibodies (IgG1 mouse anti-hum-CD31 Ab or IgG1 mouse Isotype Ab) as previously described17 using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) chemistry resulting in Ab-NP (CD31-PEG-PLA-NP and Iso-PEG-PLA-NP). NPs are flash frozen in liquid nitrogen and stored at −80C until use.
Ab attachment to nanoparticles using Mb-adapter technology
In a first step, PLA-PEG-mal NPs and PACE-PVA-VS NPs were conjugated with the Mb using the thiol reaction between the thiol reactive groups on the surface of the NPs (maleimide or vinyl sulfone) and the single terminal cysteine of the Mb. The reaction happened at room temperature with a mild mixing (Orbit Shaker, 250 rpm) for 1 h with a NPs:Mb weight ratio of 10:1. The excess of Mb was removed by centrifugation (15 min, 21,000 × g, 17 °C) and the NPs redispersed in PBS at a concentration of 5 mg/mL. In a second step, the Ab were attached to the surface of the NPs using the potent and strong binding affinity of the Mb for the Fc region of the IgG1 mouse of the Ab. The reaction happened at room temperature with a mild mixing (Orbit Shaker, 250 rpm) for 1 h with a NPs:Ab weight ratio of 18:1. The excess of Ab was removed by centrifugation (15 min, 21,000 × g, 17 °C) and the NPs redispersed in PBS at a concentration of 5 mg/mL. The resulting NP were named Ab-Mb-NP (Ab-Mb-PEG-PLA-NPs or Ab-Mb-VS-PVA-PACE-NPs). Ab-Mb-NPs were flash frozen in liquid nitrogen and stored at −80C until use.
The density of Mb and Ab on the surface of the NPs was indirectly quantified by the concentration in the supernatant from the centrifugation of each conjugation step. The quantification was made by SDS-PAGE. Successful conjugation of the antibody was verified using a cell-based binding assay.
Mb and Ab conjugation efficiency measured by SDS-PAGE
Each sample was denatured by incubation at 90 °C for 5 min in the presence of 25% (v/v) of LDS sample buffer with DTT at 1:10 (v:v) followed by a centrifugation at 4000 × g for 2 min. The samples were loaded in a 4–12% Bis-tris gel (NuPAGETM 4–12%, Bis-tris, 1.0 mm, 15 well, ThermoFisher Scientific). The first well was loaded with a standard (SeeBlueTM Plus 2 Pre-stained Protein standard, ThermoFisher Scientific). Four wells were loaded with samples of known concentration to plot a standard curve. The remaining wells were loaded with supernatant from the conjugation steps of unknown concentration. Following the electrophoresis, the gel was stained with Coomassie blue (0.1% Coomassie, 10% acetic acid, 50% methanol, 40% MilliQ water) for 1 h at room temperature under mixing. Subsequently, the gel was destained overnight under mixing. The destained solution (7.5% acetic acid, 10% ethanol, 82.5% MilliQ water) was changed every 30 min for the 2 first hours. Images of the destained gel were taken with a fluorescent scanner (ODYSSEY CLx, LI-COR). The bands intensity was analyzed with Image Studio Lite software (Li-COR Biosciences), using the same ROI and subtracting specific background for each band.
Cryo-TEM of conjugated NPs
NPs in PBS buffer were centrifuged at 21,000 × g for 15 min at 17 °C. The NPs pellet was redispersed in DI water to reach a final concentration of 5 mg/mL. Quantifoil holey carbon copper grids (Electron Microscopy Sciences) were rendered hydrophilic using a PELCO easiGlow immediately before sample preparation. NPs were vitrified using a FEI Vitrobot cryo plunger (ThermoFisher Scientific) set to 100% humidity by applying 3.5 µL of NPs suspension to the grid followed by automated removal of excess liquid with a filter paper and plunging into liquid ethane. Sample grids were kept in liquid nitrogen at all times and loaded into the ThermoFisher Glacios cryo-TEM (operating at 200 kV) equipped with a Gatan K2 Summit direct detector via the integrated autoloader. Cryo-TEM images were collected under low dose conditions at −4 µm defocus.
In vitro static cell culture NP binding assay
Human Umbilical Vein Endothelial Cells (HUVECs) were cultured in gelatin- or fibronectin-coated tissue culture plates using M199 medium supplemented with 20% (v/v) fetal bovine serum, 2% (v/v) L-glutamine, 1% (v/v) penicillin and streptomycin, and 1% (v/v) endothelial cell growth supplement. Porcine Aortic Endothelial Cells (PAECs) were cultured in gelatin- or fibronectin-coated tissue culture plate using Porcine EC Growth Medium supplemented with Porcine EC Growth Supplement. All cells were passaged a maximum of four times. Confluent cells were incubated with NPs at 50 µg/mL in the appropriate media for 2 h, then washed extensively to remove unbound NPs. For imaging purposes, cells were stained with fluorescently labeled anti-CD105 to outline the cells and/or DAPI to mark the nuclei and imaged with a fluorescent microscope (EVOS FL Auto 2, Thermo Scientific Invitrogen) or a confocal microscope (LSM 710 Duo NLO, Zeiss). For quantitative analysis, the cells were lifted with TryPLE, filtered and the intensity of fluorescence of the NPs bound to the cells was measured by flow cytometry (S1000EON, Stratodigm or Cytoflex LX, Beckman Coulter) using fluorescently labeled anti-CD31 (at 0.001 mg/mL) to draw size based gates for endothelial cells with FlowJo v10 software. The auto-fluorescence of HUVECs was subtracted from all values. The gating strategy for HUVECs in flow cytometry analysis is displayed in Supplementary Fig. 16.
In vitro cell culture under flow
An apparatus of cell culture under flow (Bioflux 200, Fluxion) was used to culture HUVECs in microfluidic channels (24-well plate two inlets one outlet, 0–20 dyne/cm2). First, the channels were coated by flowing fibronectin at 25 µg/mL in PBS for 5 min at 2 dyne/cm2 from outlet to inlets followed by 40 min at room temperature without flow. The fibronectin was washed by flowing media from outlet to inlet for 10 min at 1 dyne/cm2 and the excess of liquid was removed from all wells. Then, the cells were seeded in the channels by flowing HUVECs at 20 × 106 cells/mL for 30 s at 1 dyne/cm2 from inlet to outlet. The excess of cells in the wells was removed and replace by media in both inlets (250 µL each) and outlet (500 µL) in order to have no flow. After 12 h of incubation (37 °C, 5% CO2) the cells were confluent. Media was flown for 5 min at 1 dyne/cm2 to remove unbound cells and excess of liquid was removed from each well. Finally, NPs at 50 μg/mL in media were flown in the channel from inlet to outlet for 1 h at 0.5 dyne/cm2. Pictures were taken during this time using a fluorescent microscope (Olympus Ix71 and Olympus Ix2-UCB) equipped with a 10x objective (Olympus U Plan Fi10x-030PH1 ∞/−) and an exposure time of 0.5 s. After the hour the channel was washed from unbound NPs by flowing phenol free media containing Hoechst 33258 (1 µg/mL) for 5 min at 1 dyne/cm2. The channel was imaged under a continuous flow of 0.5 dyne/cm2 using the same fluorescent microscope equipped with a 20× objective (Japan U Plan S Apo 20×/0.75 ∞/0.17/FN26.5) and an exposure time of 0.5 s. The fluorescent intensity of the NPs bound to the cells was then quantified from the 20× images (at least 5 images/channel) using custom MATLAB code.
Ex vivo isolated vessel perfusion
An ex vivo human vessel perfusion system, as described by Lysyy et al., was used to mimic the physiological environment of blood vessels32. Briefly, de-identified human umbilical cords were obtained fresh from C-section from the Yale New Haven Hospital. The umbilical artery was carefully dissected from surrounding connective tissue and flushed with cold (4 °C) LR solution. The artery was then cut into ≈8 cm segments in length and mounted in single isolated perfusion chambers by cannulation with flushing needles. They were subsequently perfused in individual closed loops with warm complete M199 media (37 °C) at 2.5 mL/min. A bolus of a 100 µL of NPs at 5 mg/mL was injected in each loop and the perfusion was kept going for 1 h at 37 °C before being stopped. The umbilical artery segments were then recovered and rinsed with clean media. For each, whole-mount en face immunofluorescence confocal microscopy was performed with a 2 mm section and endothelial cell isolation and flow analysis were performed on the remaining section.
Endothelial cell isolation and flow cytometric analysis
For gentle endothelial cell isolation, vascular grafts were first washed with warmed PBS. The vessel was then filled with a solution of collagenase II (0.1% in PBS) and incubated for 10 min at 37 °C, and then flushed with ~1 mL of PBS + 1% BSA. The flow through containing endothelial cells was collected in a microcentrifuge tube. Cells were collected from this suspension by centrifugation for 5 min at 3500 × g, and then stained using fluorescently labeled (Alexa Fluor 488, BUV395) anti-human-endoglin (0.005 mg/mL) or an isotype control in PBS + 1% BSA for 50 min. Cells were then washed with PBS + 1% BSA and filtered. The intensity of fluorescence of the NPs bound to the cells was measured by flow cytometry (Cytoflex LX, Beckman Coulter or LSRII, BD Biosciences) and analyzed using FlowJo v10. Cytometric light filters are selected to capture the endothelial cell stain (Endoglin) as well as fluorescence (DiI) from bound NPs. The gating strategy for endothelial cells isolated from human arteries in flow cytometry analysis is displayed in Supplementary Fig. 16.
Whole-mount en face immunofluorescence confocal microscopy
Whole-mount en face immunostaining and confocal microscopy were used for visualization and evaluation of vascular endothelial layer and NP accumulation and retention. The 2 mm segment of the intact vessel was stained with ULEX-FITC (1 µg/mL) for 60 min at 4 °C. Vessel segments were then washed twice with cold PBS + 1% BSA for 10 min prior staining nuclei with Hoescht 33258 (1:10,000 dilution in PBS + 1% BSA) for 10 min. The samples were placed endothelium-side up on microscope slides, coated with a drop of pure glycerin (glycerol), and then cover slipped. To create as flat a surface as possible, silicone chemical resistant lubricant (Dow Corning, Midland, MI, USA) was applied in a perimeter around the edge of the vessel segment to hold the cover slip in place. Fluorescent images were captured with an LSM 410 spinning-disc confocal microscope and processed using Zen software (Zeiss).
Ex vivo kidney perfusion
Use of all human kidneys in this study have been approved through the research ethics of New England Donor Services US and Health Research Authority in the UK. Consent for the use of the organs for research was obtained from the donor family by local organ procurement organization representatives before organ retrieval. After in situ flushing of the abdominal organs with cold preservation solution in the donor, kidneys were retrieved and then preserved via static cold storage or hypothermic machine perfusion as described in Supplementary Table 7. Six human kidneys that were declined for transplantation were recruited into the study.
Inclusion/exclusion criteria
In order to avoid a high level of red blood cell aggregates as previously observed by Tietjen et al. and DiRito et al., we restricted the acceptable time of cold storage of the transplant-declined human kidney enrolled in the study based on the age of the donor17,41. This criterion was determined thanks to our previous experiences. The cold storage had to be <10 h for a donor >70 years old, <20 h for a donor between 50 and 70 years old and <30 h for a donor <50 years old. In addition, the organs enrolled had to be suitable for perfusion (e.g. long enough vessel to be plugged onto the machine). For these criterion, we relied upon the opinion of the transplant surgeons and the perfusion team involved in the study. Last, we observed the perfusion parameters and the macroscopic aspect of the organ during the first hour of ex vivo normothermic machine perfusion (EVNMP). Organ were only enrolled for nanoparticles injection if the kidney was perfusing well which was characterized by a homogeneous pink-red color, no spike in resistance, a urine output >100 mL/h/100 g of kidney and a stable blood flow ~50 mL/min/100 g of kidney. With these inclusion/exclusion criteria only one kidney was excluded due to bad perfusion (K1). The other 6 consecutive organs offered fitted all the criteria and were injected with nanoparticles.
Normothermic machine perfusion
Upon arrival to the laboratory, kidneys were weighed and prepared for perfusion through careful isolation of the renal artery, vein and ureter. The renal artery was cannulated with a 14fr catheter. The ureter was cannulated with a 10fr catheter. Kidneys were flushed with 500 mL of cold Ringer’s solution (Baxter Healthcare) prior to perfusion. As previously described, the kidney perfusion circuit utilized pediatric cardiopulmonary bypass technology (Medtronic) and consisted of a centrifugal blood pump (Bio-Pump 560), a heat exchanger (Chalice Medical), a venous reservoir (Medtronic), 1/4-inch polyvinyl chloride tubing, and a Pixie membrane oxygenator (Medtronic)41. The hardware included a speed controller and a TX50P flow transducer. The circuit was primed with Ringer’s solution and one unit of AB packed red cells (plasma-free) from the New York Blood Center, or blood compatible units of red cells for the kidneys perfused in the UK. 25 mL of 10% mannitol, 8 mg of dexamethasone, 3 mL of heparin (1000 IU/ml), 25 mL of 8.4% sodium bicarbonate, and 10 mL of 5% glucose were added to the perfusate. Ringer’s solution was used to replace urine output milliliter-for-milliliter. The perfusate was circulated from the venous reservoir through the centrifugal pump at 1450 rpm into the membrane oxygenator, where it was oxygenated and also warmed to 38 °C. It then flowed through the arterial limb of the circuit to the renal artery. Venous return from the renal vein was fed back into the reservoir. Kidneys were placed on the circuit and perfused for an initial assessment period of 60 min. If the perfusionist deemed that the kidney was stable and functioning well at the end of that 60-min period, the experiment would proceed with the NPs injection. A bolus of 10 mL of NPs suspension containing non targeted NPs (DiO loaded) and Ab-Mb-NPs (anti-CD31 or Isotype, DiI loaded) both at 5 mg/mL would be delivered into the arterial port of the perfusion circuit reaching a final circulating concentration of 50 µg/mL in the perfusate. The perfusionist was blinded to what NPs they were delivering in each kidney for the four last kidneys (two pairs) enrolled. The perfusion was continued for an additional 4 h after NPs injection.
Sample collection and processing
Renal blood flow (RBF) and total urine output was continuously monitored and registered throughout the NMP procedure. Perfusate and urine samples collected throughout the perfusion were snap frozen and stored at −80 °C. Wedge biopsies were collected prior to the start of perfusion and before NPs injection at 1 h of NMP. At the end of the 5-h procedure kidneys were flushed with LR, bisected and three biopsies spanning the cortex and medulla were collected (one in the upper part, one in the middle and one in the lower part). Biopsies were snap-frozen in liquid nitrogen upon collection and stored at −80 °C until further analysis. From each post perfusion biopsy and from the biopsy before NPs, three tissue sections 100 µm apart in depth were stained with Ulex europaeus Agglutinin I (ULEX, a lectin which can be used to identify endothelial cells), and whole sections of tissue (Fig. 5a, b) were imaged at 20x resolution and tiled (200 to 350 images per section) using a fluorescent microscope EVOS FL Auto 2 (Thermo Scientific Invitrogen). Filter cubes with the following parameters were used for each channel: blue channel with 357/44 nm Excitation; 447/60 nm Emission, green channel with 482/25 nm Excitation; 524/24 nm Emission, red channel with 531/40 nm Excitation; 593/40 nm Emission, and far red with 628/40 nm Excitation; 692/40 nm Emission. A consistent laser power (50% laser power) and exposure were used for all image collection (0.2 s exposure for green and 0.15 s exposure for red, 0.05 s exposure for far red).
Image analysis
Images were analyzed using custom MATLAB code which is available upon request and is described in Supplementary Fig. 8. Briefly, the background signal in both the green channel and the red channel was measured from biopsy taken before NPs were introduced for each kidney. Using an average of the values obtained from ~900 images, a value was calculated that would make 98% of the image black if subtracted from each pixel. This value was around 400 for green images and 150 for red images. Next, a vascular binary mask was built based on positive ULEX staining in the far red channel. Briefly, each image in the far red channel was analyzed using an adjusted Otsu’s method to determine a threshold that when applied, resulted in an accurate area representation of the vasculature in each image. This value varied between 600 and 1200 based on the particular kidney and on the region of tissue (cortex vs medulla). The determined threshold value was applied to the far red image, resulting in a binary mask (value of 1 where vasculature is present, value of 0 where it is not). This mask was applied by multiplication to each of the green and red channel images, to remove signal from tissue regions not contained inside the vasculature. Next, an inverse binary mask was built from positive green (nonspecific) NP signal using a threshold of 900, which was predetermined to accurately represent green NP accumulation area. This mask (value of 1 where no NPs were found, value of 0 where green NPs were found) was applied by multiplication to the red channel image, to remove areas of green and red NP colocalization. Finally, the corrected red channel image was binarized (using a threshold of 400, and the sum of nonzero elements per image was calculated. The cumulative sum of all nonzero elements in the red channel per slice (~300 images) was then divided by the cumulative sum of nonzero elements in the vascular channel minus the cumulative sum of nonzero elements in the green channel (vascular mask applied) per slice to obtain a percent of available vascular area covered by NPs.
Statistical analyses
The results of the experiments are expressed as the means ± the standard deviation. Statistical analyses were noted were performed by ANOVA and a post hoc Tukey’s Test where appropriate for comparison between groups using Prism 8.0 (GraphPad Software, Inc.). In other cases, where noted, statistical analysis was performed with multiple T-tests with a Bonferoni correction using Prism 8.0 (GraphPad Software, Inc.). A value of p < 0.05 was considered statistically significant. Exact p values can be found in the source data file.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
