

Abstract
The miniaturization of ferroelectric devices in non-volatile memories requires the device to maintain stable switching behavior as the thickness scales down to nanometer scale, which requires the coercive field to be sufficiently large. Recently discovered metal-free perovskites exhibit advantages such as structural tunability and solution-processability, but they are disadvantaged by a lower coercive field compared to inorganic perovskites. Herein, we demonstrate that the coercive field (110 kV/cm) in metal-free ferroelectric perovskite MDABCO-NH4-(PF6)3 (MDABCO = N-methyl-N’-diazabicyclo[2.2.2]octonium) is one order larger than MDABCO-NH4-I3 (12 kV/cm) owing to the stronger intermolecular hydrogen bonding in the former. Using isotope experiments, the ferroelectric-to-paraelectric phase transition temperature and coercive field are verified to be strongly influenced by hydrogen bonds. Our work highlights that the coercive field of organic ferroelectrics can be tailored by tuning the strength of hydrogen bonding.
Similar content being viewed by others
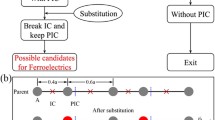

Introduction
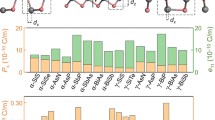
Since the discovery of ferroelectricity in 1920 in Rochelle salt, enormous research interests have been dedicated to the study of ferroelectrics41,42. We constructed a √2 × √2 × 1 supercell based on the unit cell of the ferroelectric phase, and built the centrosymmetric reference phase via rotation, displacement and distortion of the components, after which two MDABCO molecules in the simulation cell are aligned antiparallel to each other (Fig. 5a). The calculated polarization of MNP3 is shown in Fig. 5b, where it varies continuously from 0 to 6.5 μC/cm2 along the dynamic path, thus the spontaneous polarization agrees well with the experimental value of 5.7 μC/cm2. Although the larger displacement of MDABCO in MNP3 than MNI3 (Fig. 2d) gives a larger dipole moment in the former, the larger unit cell volume of MNP3 than MNI3 offsets the increased polarization as the overall polarization is defined by the sum of dipole moment per unit volume. Furthermore, in MNI3, there is a large polarization contribution from the off-centre displacement of NH4+ at B site (δB = 0.6366 pm) because of its larger rB/rX ratio (0.664) compared to the typical octahedron ratio (0.414–0.592) predicted by Pauling’s rule. On the other hand, the NH4+ ion in MNP3 is located at the centre of the octahedron (δB = 0.0312 pm) with a rB/rX ratio of 0.570, thus there is no off-centre displacement.

a The dynamic path of ferroelectric phase transition in MNP3. b The polarization value of MNP3 was obtained by Berry phase calculations. The path connects the centrosymmetric reference phase (λ = 0) to the ferroelectric phase (λ = 1). c Electrostatic potential of MNI3 and MNP3, and the Mulliken charges of H atoms participating in hydrogen bonds.
The displacement of MDABCO in the polarization direction with respect to the MNP3 framework is the primary driver of the ferroelectricity of MNP3. The large electrostatic potential differences between MDABCO-NH and PF6 increase the hydrogen-bond strength compared to MNI3. Mulliken charge calculations for MNP3 and MNI3 reveal that PF6− has a more pronounced inductive effect than I− on the H atom in MDABCO (Fig. 5c), thus the stronger N–H…F hydrogen retards the polarity reversal process of organic ferroelectric perovskites. Therefore, any reinforcement of hydrogen bonding will hinder the ferroelectric switching process, since multiple hydrogen bonds need to be successively broken and established during the rotation of MDABCO, as illustrated in Fig. 5a. This is the root cause of the enhanced coercive field for MNP3.
In conclusion, incorporating stronger hydrogen bonds in MDABCO-NH4-I3 by the substitution of PF6− for I− increase the coercive field by an order of magnitude from ~12 kV/cm to 110 kV/cm. This enables a threshold voltage of 1 V for a 90 nm thick film, which complies with the miniaturization requirement of FeRAM devices. Both experimental results and DFT calculations provide robust evidence for the mechanism of MDABCO rotation upon ferroelectric switching and phase transition, the dynamics of which are modulated by the intermolecular hydrogen bonding interactions between MDABCO2+ and PF6−. This work demonstrates that regulating intermolecular interactions between the cation and anion can be used for the engineering of the coercive field in organic ferroelectric materials.
Methods
Synthesis
Detailed synthetic methods for preparing precursors are provided in Supplementary Information. For growing MNP3 crystal, 209 mg (0.5 mmol) of MDABCO-(PF6)2 and 81.5 mg (0.5 mmol) of NH4PF6 were dissolved in acetonitrile until saturated, and the parallelogrammic single-crystals were grown after slow evaporation.
Characterization
All chemicals were purchased from Sigma–Aldrich without further purification. 1H and 13C nuclear magnetic resonance (NMR) was taken by AVII 400 MHz NMR spectrometer of Bruker. Thermogravimetric analyses (TGA) were performed under a nitrogen atmosphere with a heating rate of 10 °C/min using a TA Instruments Trios V3.1 thermogravimetric analyzer. Differential scanning calorimetry (DSC) scans were performed under a nitrogen atmosphere with a heating rate of 10 °C/min using Mettler-Toledo DSC. The dielectric constant was measured by CVU unit in Keithley-SCS4200 with the pelleted sample. Powder X-ray diffraction (PXRD) patterns were recorded on a Bruker D8 Focus Powder X-ray diffractometer using Cu Kα radiation (40 kV, 40 mA) at room temperature. Ferroelectric P-E curve was measured with Precision Multiferroic II Ferroelectric Test System of the Radiant Technologies with high voltage amplifier. Piezoresponse force microscopy (PFM) tests were performed on Bruker Dimension Icon Atomic Force Microscope with grown crystals or spray-coated samples on ITO.
Density functional theory (DFT) calculations
We performed the Berry phase calculations41,42 within the DFT framework as implemented in the Vienna ab initio simulation package (VASP)43,44. The exchange-correlation interactions were treated within the Perdew-Burke-Ernzerh (PBE) generalized gradient approximation45. To complement the deficiencies of DFT in treating dispersion interactions, the third-generation (D3) van der Waals corrections proposed by Grimme46 were employed. The plane-wave cutoff energy was set to 520 eV, and the k-point mesh to 3 × 3 × 4. The polarization was calculated using a supercell twice the size of the unit cell so that a centrosymmetric reference phase can be constructed. A convergence threshold of 0.01 eV/Å in force was reached in structural optimization. Electrostatic potential and Mulliken charge were calculated by DMol347,48 code in Materials Studio using a double numerical polarized basis set and PBE45 exchange-correlation functional.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated and analyzed in this study are included in the Article and its Supplementary Information, and are also available from corresponding authors upon request. Crystallographic data for this paper can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. CCDC- 2085249 (MNP3 at RT) and CCDC- 2085250 (D-MNP3 at RT).
References
Whatmore, R. W., You, Y.-M., **ong, R.-G. & Eom, C.-B. 100 years of ferroelectricity—a celebration. APL Mater. 9, 070401 (2021).
Peña, M. A. & Fierro, J. L. G. Chemical structures and performance of perovskite oxides. Chem. Rev. 101, 1981–2018 (2001).
Mischenko, A. S., Zhang, Q., Scott, J. F., Whatmore, R. W. & Mathur, N. D. Giant electrocaloric effect in thin-film PbZr0.95Ti0.05O3. Science 311, 1270–1271 (2006).
Horiuchi, S. & Tokura, Y. Organic ferroelectrics. Nat. Mater. 7, 357–366 (2008).
Li, W. et al. Chemically diverse and multifunctional hybrid organic–inorganic perovskites. Nat. Rev. Mater. 2, 16099 (2017).
Wei, L., Stroppa, A., Wang, Z. M. & Gao, S. In Hybrid Organic-Inorganic Perovskites. (Wiley-VCH, 2020).
Song, X., Hodes, G., Zhao, K. & Liu, S. Metal-free organic halide perovskite: a new class for next optoelectronic generation devices. Adv. Energy Mater. 11, 2003331 (2021).
Li, K. et al. Electronic structures and elastic properties of a family of metal-free perovskites. Mater. Chem. Front. 3, 1678–1685 (2019).
Bremner, C. A., Simpson, M. & Harrison, W. T. A. New molecular perovskites: cubic C4N2H12·NH4Cl3·H2O and 2-H hexagonal C6N2H14·NH4Cl3. J. Am. Chem. Soc. 124, 10960 (2002).
Hang, T., Zhang, W., Ye, H.-Y. & **ong, R.-G. Metal–organic complex ferroelectrics. Chem. Soc. Rev. 40, 3577–3598 (2011).
Shi, P.-P. et al. Symmetry breaking in molecular ferroelectrics. Chem. Soc. Rev. 45, 3811–3827 (2016).
Zhang, W. & **ong, R.-G. Ferroelectric metal–organic frameworks. Chem. Rev. 112, 1163–1195 (2012).
Zhang, H.-Y., Tang, Y.-Y., Shi, P.-P. & **ong, R.-G. Toward the targeted design of molecular ferroelectrics: modifying molecular symmetries and homochirality. Acc. Chem. Res. 52, 1928–1938 (2019).
Ye, H.-Y. et al. Metal-free three-dimensional perovskite ferroelectrics. Science 361, 151 (2018).
Wang, J.-J., Fortino, D., Wang, B., Zhao, X. & Chen, L.-Q. Extraordinarily large electrocaloric strength of metal-free perovskites. Adv. Mater. 32, 1906224 (2020).
Mikolajick, T. et al. Next generation ferroelectric materials for semiconductor process integration and their applications. J. Appl. Phys. 129, 100901 (2021).
Park, M. H. et al. Ferroelectricity and antiferroelectricity of doped thin HfO2-based films. Adv. Mater. 27, 1811–1831 (2015).
Fan, Z., Chen, J. & Wang, J. Ferroelectric HfO2-based materials for next-generation ferroelectric memories. J. Adv. Dielectr. 06, 1630003 (2016).
Samara, G. A. The effects of deuteration on the static ferroelectric properties of KH2PO4 (KDP)†. Ferroelectrics 5, 25–37 (1973).
Nayeem, J., Wakabayashi, H., Kikuta, T., Yamazaki, T. & Nakatani, N. Ferroelectric properties of deuterated glycine phosphite. Ferroelectrics 269, 153–158 (2002).
Horiuchi, S., Kumai, R. & Tokura, Y. A supramolecular ferroelectric realized by collective proton transfer. Angew. Chem. Int. Ed. 46, 3497–3501 (2007).
Ubbelohde, A. R. & Gallagher, K. J. Acid-base effects in hydrogen bonds in crystals. Acta Crystallogr. 8, 71–83 (1955).
Zhang, W.-Y. et al. Precise molecular design of high-Tc 3D organic–inorganic perovskite ferroelectric: [MeHdabco]RbI3 (MeHdabco = N-Methyl-1,4-diazoniabicyclo[2.2.2]octane). J. Am. Chem. Soc. 139, 10897–10902 (2017).
Loiacono, G. M., Balascio, J. F. & Osborne, W. Effect of deuteration on the ferroelectric transition temperature and the distribution coefficient of deuterium in K(H1−xDx)2PO4. Appl. Phys. Lett. 24, 455–456 (1974).
Shi, C., Zhang, X., Yu, C.-H., Yao, Y.-F. & Zhang, W. Geometric isotope effect of deuteration in a hydrogen-bonded host–guest crystal. Nat. Commun. 9, 481 (2018).
Horiuchi, S., Ishibashi, S., Kobayashi, K. & Kumai, R. Coexistence of normal and inverse deuterium isotope effects in a phase-transition sequence of organic ferroelectrics. RSC Adv. 9, 39662–39673 (2019).
Ai, Y., Lv, H.-P., Wang, Z.-X., Liao, W.-Q. & **ong, R.-G. H/F substitution for advanced molecular ferroelectrics. Trends Chem. 3, 1088–1099 (2021).
Whatmore, R. In Springer Handbook of Electronic and Photonic Materials. (Springer International Publishing, 2017).
Harris, R. K. & Olivieri, A. C. Quadrupolar effects transferred to spin-12 magic-angle spinning spectra of solids. Prog. Nucl. Magn. Reson. Spectrosc. 24, 435–456 (1992).
Gobetto, R. et al. 1H MAS, 15N CPMAS, and DFT investigation of hydrogen-bonded supramolecular adducts between the diamine 1,4-diazabicyclo-[2.2.2]octane and dicarboxylic acids of variable chain length. Chem. Mater. 17, 1457–1466 (2005).
Harada, J. et al. Directionally tunable and mechanically deformable ferroelectric crystals from rotating polar globular ionic molecules. Nat. Chem. 8, 946–952 (2016).
Harada, J. et al. Ferroelectricity and piezoelectricity in free-standing polycrystalline films of plastic crystals. J. Am. Chem. Soc. 140, 346–354 (2018).
Harada, J. et al. Plastic/ferroelectric crystals with easily switchable polarization: low-voltage operation, unprecedentedly high pyroelectric performance, and large piezoelectric effect in polycrystalline forms. J. Am. Chem. Soc. 141, 9349–9357 (2019).
Harada, J. Plastic/ferroelectric molecular crystals: ferroelectric performance in bulk polycrystalline forms. APL Mater. 9, 020901 (2021).
Das, S., Mondal, A. & Reddy, C. M. Harnessing molecular rotations in plastic crystals: a holistic view for crystal engineering of adaptive soft materials. Chem. Soc. Rev. 49, 8878–8896 (2020).
Stephan, K. & Peter, L. Ferroelectric polarization in multiferroics. Phys. Sci. Rev. 4, 20190015 (2019).
Song, X.-J. et al. Bistable state of protons for low-voltage memories. J. Am. Chem. Soc. 142, 9000–9006 (2020).
Foster, C. M. et al. Single-crystal Pb(ZrxTi1−x)O3 thin films prepared by metal-organic chemical vapor deposition: Systematic compositional variation of electronic and optical properties. J. Appl. Phys. 81, 2349–2357 (1997).
Chen, X., Han, X. & Shen, Q.-D. PVDF-based ferroelectric polymers in modern flexible electronics. Adv. Electron. Mater. 3, 1600460 (2017).
Aizu, K. Possible species of “Ferroelastic” crystals and of simultaneously ferroelectric and ferroelastic crystals. J. Phys. Soc. Jpn. 27, 387–396 (1969).
Vanderbilt, D. & King-Smith, R. D. Electric polarization as a bulk quantity and its relation to surface charge. Phys. Rev. B 48, 4442–4455 (1993).
King-Smith, R. D. & Vanderbilt, D. Theory of polarization of crystalline solids. Phys. Rev. B 47, 1651–1654 (1993).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92, 508–517 (1990).
Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 113, 7756–7764 (2000).
Acknowledgements
K.P.L. would like to acknowledge the Singapore’s Ministry of Education Tier 2 grant (MOE2019-T2-1-037), the Shenzhen Peacock Plan (Grant No. KQTD2016053112042971). F.P. would like to acknowledge the Guangdong Special Support Program, and Soft Science Research Project of Guangdong Province (No. 2017B030301013). K.Y. would like to acknowledge partial supports by A*STAR, Singapore, RIE2020 Advanced Manufacturing and Engineering (AME) Programmatic Fund, (Grant No. A20G9b0135).
Author information
Authors and Affiliations
Contributions
H.S.C. designed, synthesized and characterized the MNP3 crystals. K.P.L. supervised the research. S.L., S.Z. and F.P. did DFT calculations. I.H.P. did a single-crystal XRD structure analysis. W.H.L. and K.Y. did a ferroelectric experiment and analysis. Z.Z. and Q.H.X. did a temperature-dependent SHG experiment. K.C.K. did PFM measurement. L.W. did temperature-dependent dielectric measurement. I.H.O. did an analysis on the deuteration effect. H.S.C., S.L. and K.P.L. wrote the manuscript in consultation with all authors. H.S.C. and S.L. contributed equally.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, H.S., Li, S., Park, IH. et al. Tailoring the coercive field in ferroelectric metal-free perovskites by hydrogen bonding. Nat Commun 13, 794 (2022). https://doi.org/10.1038/s41467-022-28314-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28314-8
- Springer Nature Limited
This article is cited by
-
Chiral multiferroicity in two-dimensional hybrid organic-inorganic perovskites
Nature Communications (2024)