

Abstract
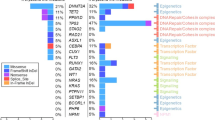
Multiple myeloma (MM) is a plasma-cell neoplasm that is treated with high-dose chemotherapy, autologous stem cell transplant (ASCT) and long-term immunomodulatory drug (IMiD) maintenance. The presence of somatic mutations in the peripheral blood is termed clonal hematopoiesis of indeterminate potential (CHIP) and is associated with adverse outcomes. Targeted sequencing of the stem cell product from 629 MM patients treated by ASCT at the Dana-Farber Cancer Institute (2003–2011) detects CHIP in 136/629 patients (21.6%). The most commonly mutated genes are DNMT3A, TET2, TP53, ASXL1 and PPM1D. Twenty-one from fifty-six patients (3.3%) receiving first-line IMiD maintenance develop a therapy-related myeloid neoplasm (TMN). However, regardless of CHIP status, the use of IMiD maintenance associates with improved PFS and OS. In those not receiving IMiD maintenance, CHIP is associated with decreased overall survival (OS) (HR:1.34, p = 0.02) and progression free survival (PFS) (HR:1.45, p < 0.001) due to an increase in MM progression.
Similar content being viewed by others


Introduction
Multiple myeloma (MM) is an incurable clonal plasma-cell malignancy that is more common in older individuals and accounts for approximately 10% of all hematological malignancies1. The standard of care for fit newly diagnosed patients in the United States is induction chemotherapy with a combination of a proteasome inhibitor and immunomodulatory drug (IMiD) followed by high dose melphalan chemotherapy and autologous stem cell transplantation (ASCT). This is typically followed by IMiD maintenance until disease progression. Not all patients benefit from ASCT and it can be associated with significant long-term toxicities including cytopenias and therapy-related myeloid neoplasms (TMN), a risk that is further increased by the use of lenalidomide maintenance2,3,4. However, the clinical benefit of lenalidomide maintenance as seen in improved overall survival (OS) clearly outweigh its risk. A better understanding of which patients may benefit from ASCT and maintenance therapy is an important question and active area of ongoing clinical investigation.
A number of recent studies have identified recurrent somatic mutations in the blood of otherwise healthy adults, a condition referred to as clonal hematopoiesis of indeterminate potential (CHIP)5. CHIP is associated with a 0.5–1% risk of progression to a non-plasma-cell hematologic neoplasm, in particular myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML)19, the presence of CHIP prior to ASCT was not associated with an increased risk of TMN (p = 0.4). However, IMiD maintenance was significantly associated with develo** a subsequent TMN (p = 0.047) (Fig. 2a, b) and the presence of CHIP did not increase the risk among those receiving first-line IMiD maintenance or IMiD maintenance at any point post-ASCT (Fig. 2c, d). Nine of the 21 patients who developed MDS/AML were still alive at time of analysis. Seven patients had a myeloma relapse before the diagnosis of MDS/AML. Of the 12 patients who died, 5 passed away from the TMN while 7 died from a combination of myeloma progression and MDS/AML.

a Cumulative incidence of MDS/AML in patients with IMiD maintenance versus patients without maintenance, with death as an absorbing competing risk. Cumulative incidence of develo** MDS/AML among patients with CHIP vs. no CHIP, with death as an absorbing competing risk, b among all patients, c patients receiving first-line IMiD maintenance and d patients receiving IMiDs at any point post ASCT. Groups were tested for equality using a two-sided Gray’s test to compare subdistributions for each competing risk.
We next asked whether CHIP at the time of ASCT was clonally related to a subsequent MDS or AML. Sequential samples from 14 of the 21 TMN patients were available for targeted sequencing (Supplementary Tables 5 and 6). 10/14 patients had an identifiable driver mutation at the time of TMN diagnosis, most commonly in TP53, but only 4 of those patients had a detectable mutation at the time of ASCT. Among the 6 patients in whom we did not initially find CHIP, we were able to identify the TMN driver mutation in 4 of the ASCT samples at VAFs below our threshold of 0.01. Thus, in 8 of 10 patients with identifiable mutations at the time of TMN diagnosis, at least one somatic mutation could also be detected prior to ASCT. In most cases, the driver mutation present prior to ASCT had expanded at the time of TMN diagnosis (Supplementary Table 6). It is possible that with deeper sequencing pre-existing mutations could be found in these additional 2 TMN cases as well.
CHIP is associated with adverse outcomes
Having found no evidence for higher TMN risk in patients with CHIP, we examined whether CHIP was associated with other adverse outcomes. Out of 629 patients in our cohort, 376 patients had died at the time of analysis. The median OS and progression-free survival (PFS) of our cohort were 7.1 and 2.5 years, respectively. After stratification based on age, International Staging System (ISS) and number of treatment lines prior to ASCT, we modeled the association of CHIP with OS and PFS. The median OS of patients with CHIP was 5.3 years, significantly lower than in those without CHIP (7.5 years) (HR: 1.34, p = 0.02, stratified multivariable cox regression model) (Fig. 3a). Interestingly, CHIP was also associated with a lower median PFS of 2.2 years compared to 2.6 years in those without CHIP (HR: 1.45, p < 0.001, stratified multivariable cox regression model) (Fig. 3b). We also observed a worse OS and PFS in patients carrying more than one mutation (Supplementary Fig. 5).

a OS and b PFS models for all 629 patients after stratifying by age, ISS and number of lines of therapy prior to ASCT to investigate the effect of CHIP and IMiD maintenance on outcome. Two-sided Wald p-values are shown for each model coefficient with significant effects displayed in red. Exact p-values: A: 0.0197276361 and 0.0000692041; B: 0.0007310355 and 1.547737 × 10−15. HR: Hazard Ratio; LCI: Lower Confidence Interval; UCI: Upper Confidence Interval.
Unlike prior studies of CHIP, we did not observe excess deaths related to cardiovascular disease or stroke, possibly due to the aggressive nature of multiple myeloma8,19. The most common cause of death was myeloma disease progression followed by respiratory failure and sepsis. Interestingly, patients with CHIP responded less well to induction therapy such that CHIP was associated with a higher post-induction median level of β2-microglobulin (2.3 mg/dL in those with CHIP compared to 2.0 mg/dL in those without [p = 0.008]), and a smaller percentage decrease in M-spike level (p = 0.008) post-induction as compared to diagnosis (Supplementary Tables 7–9).
IMiD maintenance is associated with improved outcomes in all patients
Treatment with IMiDs has been reported to increase the risk of secondary malignancies including MDS and AML2,3,4. Almost all patients received thalidomide or lenalidomide at some point throughout the course of their disease. Therefore, we asked whether treatment with IMiDs was associated with adverse outcomes in patients with CHIP. Only 57% of the patients in our cohort received first-line IMiD maintenance, with 22% receiving it for at least 3 years (range: 0.1–14.9). First-line IMiD maintenance was associated with a longer median OS of 8.5 years compared to 5.6 years in those not receiving IMiD maintenance [HR: 0.65 (0.52–0.80), p < 0.001] (Fig. 3a). As expected, IMiD maintenance was associated with a longer PFS of 3.4 years compared to 1.5 years in those not receiving IMiD maintenance [HR: 0.47 (0.39–0.56), p < 0.001] (Fig. 3b). Only 16 patients received proteasome inhibitor-based maintenance, too few to draw significant conclusions (Supplementary Fig. 6).
In patients not receiving IMiD maintenance, CHIP was associated with a significantly lower median OS of 3.6 years compared to 6.6 years in those who did not have CHIP mutations (p = 0.013, stratified analysis). However, there was no significant difference in those who received maintenance (median OS of 7.7 and 8.9 years for CHIP and no CHIP, respectively, p = 0.49, stratified analysis) (Fig. 4a). Similarly, in patients not receiving IMiD maintenance, CHIP was associated with a significantly lower median PFS of 1.1 years compared to 1.8 years (p < 0.001, stratified analysis). There was also no difference in PFS among patients with and without CHIP who received IMiD maintenance (median PFS of 3.3 and 3.6 years for CHIP and no CHIP, respectively, p = 0.59, stratified analysis) (Fig. 4b). These results suggest the presence of an interaction between the effect of having CHIP and IMiD maintenance, which is significant when it comes to PFS outcome [HR: 0.51 (0.34–0.79), p = 0.002] (Supplementary Fig. 7).

a OS and b PFS among patients with CHIP versus those without CHIP in the context of receiving versus not receiving IMiD maintenance post ASCT. Overall and pairwise two-sided log-rank p-values are shown unadjusted for multiple testing.
We next asked whether mutations in specific genes were associated with worse outcomes. The two most commonly mutated genes were DNMT3A and TET2, which were associated with a significantly reduced PFS and OS as compared to patients without CHIP in the absence of IMiD maintenance (Supplementary Fig. 8). In particular, patients with the p.R882 DNMT3A mutation had the worst median OS of 1 year (p = 0.008, stratified analysis) and median PFS of 0.9 years (p = 0.007, stratified analysis) compared to patients without CHIP (Supplementary Fig. 9). However, in patients who received IMiD maintenance, the decrease in OS and PFS seen in p.R882 patients was completely abrogated.
Altogether, these data suggest that the presence of CHIP at time of ASCT does not increase the risk of TMN associated with IMiD maintenance and that patients with CHIP, when treated with IMiD maintenance, obtain a survival benefit similar to that seen in MM patients generally.
Discussion
Here we report the first study to date examining the relationship between CHIP and clinical outcomes in MM, involving over 600 patients undergoing ASCT at a single center with a median follow-up of 9.7 years. The frequency of CHIP in this cohort is lower than that seen in patients with relapsed NHL undergoing ASCT and had a mutational spectrum more similar to that seen in healthy adults7,8,19. The differences observed between CHIP in MM and NHL are potentially related to the shorter duration of chemotherapy exposure in patients receiving induction therapy for MM and less use of DNA damaging agents that may select for specific mutant clones. The mutations seen most frequently in patients with relapsed NHL are found in PPM1D and TP53, two genes known to play important roles in the response to DNA damage and chemotherapy resistance30,31,32.
TMN is one of the most feared complications of treatment for MM and ASCT, in particular, and the presence of mutant hematopoietic clones has been proposed to herald its development. However, we did not observe an increased risk of TMN in patients with CHIP undergoing ASCT. This may be because the mutations selected for by MM induction therapy do not carry a high risk of subsequent myeloid malignancy14,15. Consistent with this hypothesis, the most common mutations in those who developed a TMN were in TP53, which was found relatively infrequently (2.9%) in our cohort at time of ASCT. Further prospective studies are needed to determine whether the presence of TP53 or other specific mutations at the time of ASCT carry a high risk of TMN development and thus might warrant avoidance of ASCT in MM patients harboring them.
We detected CHIP in 21.6% of MM patients at the time of ASCT and found it to be associated with both decreased OS and PFS. In contrast to previous reports, this was not due to an increased risk of TMN or cardiovascular disease19. Unlike in NHL where ASCT is a potentially curative therapy and many patients may expect a long-life expectancy after transplant, MM almost uniformly relapses and may provide insufficient time for cardiovascular disease to manifest. As treatment for MM continues to improve with a concomitant increase in patient life expectancy the potential long-term non-hematopoietic risks associated with CHIP will need to be evaluated further.
Surprisingly, the primary effect of CHIP on survival was due to an increased risk for myeloma progression. Consistent with this finding, patients with myeloma and CHIP had a higher level of β2-microglobulin and a smaller percentage decrease in their M-protein post induction, compared to those without CHIP. There are several potential mechanisms by which CHIP could promote increased progression of MM. It is possible that patients with CHIP are more prone to the development of cytopenias and other toxicity from therapy, thus increasing the frequency of treatment delays or dose reductions and limiting their ability to receive optimal myeloma directed therapy. Alternatively, the presence of CHIP could alter the bone marrow (BM) microenvironment in such a way as to promote MM progression. Myeloid cells carrying mutations in TET2 and DNMT3A have been reported to stimulate inflammation through upregulation of IL-1β and IL-611,33. Whether a hyperinflammatory phenotype within the BM niche might favor the growth of MM cells and promote more aggressive disease is a prospect that will require further investigation in both animal models and patients.
IMiD maintenance has demonstrated a clear survival advantage in the post-ASCT setting2,3,4. However, it has also been associated with an increased risk of TMN and mutations in TP53 have been reported to promote clonal expansion and the development of resistance in del(5q) MDS treated with lenalidomide34,35. While induction with lenalidomide could have led to the relative enrichment of TP53 at time of ASCT, we surprisingly saw no increase in TMNs in patients with CHIP and the use of IMiD maintenance. In fact, IMiD maintenance was not only associated with an improvement in both PFS and OS but it completely abrogated the deleterious effects of CHIP in the post-ASCT setting. The findings from this study are limited by the fact that it is retrospective and includes patients treated during the introduction of IMiD maintenance and thus not all patients received maintenance therapy. Follow-up studies examining the large randomized studies of placebo vs. lenalidomide maintenance could further define the role of IMiDs in the survival of patients with CHIP. In addition, because all patients received ASCT, the role of high dose melphalan and stem cell transplant cannot be fully dissected in this study and warrants further investigation within trials comparing clinical outcomes of upfront vs. delayed/salvage ASCT or transplant-based vs. drug-based consolidation36,37,38,39,40.
In summary, we found CHIP to be a common entity among MM patients undergoing ASCT. The presence of CHIP was associated with worse outcomes and thus, it would be tempting to screen newly diagnosed MM patients for CHIP before ASCT. However, our data suggest that ASCT, when followed by IMiD maintenance, can be safely utilized regardless of CHIP status. Further well-controlled prospective clinical trials are needed to investigate the interaction between CHIP, transplant and IMiDs on outcomes in multiple myeloma.
Methods
Cohort
Following institutional review board (IRB) approval, we collected the clinical data and all available cryopreserved products of mobilized autologous stem-cells from 629 MM patients who underwent ASCT between January 2003 and December 2011 at the Dana-Farber Cancer Institute (DFCI) in Boston, MA. The cutoff date of 2011 was used to enable enough years of follow up following stem cell transplantation and allow for the monitoring of TMNs and survival data. Clinical information was collected through November 2019. The study design complied with the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice. All subjects previously provided written informed consent to allow the collection of clinical information and genetic analysis of PB and BM samples for research purposes (DF/HCC IRB 01–206, 07–150 and 16–529).
While all 629 patients received ASCT, 21 of those patients received tandem ASCT, and three received a second ASCT at a later time point. Also, 38 patients received allogenic stem cell transplant, seven of which were tandem, and the rest got them at a later time point post relapse.
Genomic studies
Deep targeted sequencing was performed on the stem-cell products of 629 MM patients, as well as on available samples of PB and BM aspirates obtained from 15 patients at the time of pre-mobilization and when they developed a hematologic second primary malignancy post-ASCT, respectively. A custom target bait panel of 224 genes was used, including, pan-cancer, myeloma and myeloid malignancy-associated genes (Agilent SureSelectXT hybrid capture system). See Supplementary Table 1 for a list of genes and coordinates. Libraries for the stem cell products were constructed automatically, using the Agilent Bravo robot, and were sequenced on the Illumina HiSeq 4000 platform in pools of 32 samples, achieving a 978X total depth of coverage. Libraries of PB and BM samples were constructed manually and were sequenced on the Illumina HiSeq 2500 platform in pools of 24, achieving 556X total depth of coverage. To detect large-scale copy number alterations that reflect tumor cells within the stem cell products, we also performed ULP-WGS at an average genome-wide fold coverage of 0.1×. Detailed information on library preparation, sequencing platforms and computational analysis for targeted sequencing and ULP-WGS is provided in the supplementary information.
Sequencing was done at the Broad Institute, Cambridge, MA, USA.
Computational analyses
Sequencing data was analyzed using the pipelines of the Broad Institute of Harvard and MIT (Firehose, www.broadinstitute.org/cancer/cga). To estimate the presence of tumor, we performed ultra-low pass whole-genome sequencing (ULP-WGS) of all samples to an average genome-wide fold coverage of 0.1×41. The depth of coverage was determined using ichorCNA41,42,43, to estimate large-scale copy number alterations (CNAs) and the fraction of tumor in ULP-WGS. Low coverage samples (<0.05×) were manually reviewed to determine tumor fraction. All samples had a low tumor fraction (3–5.4%).
The targeted sequencing data of our samples were aligned using BWA-mem and the base qualities of the aligned data were re-calibrated using GATK3 Base Quality Score Recalibration (BQSR)44,45. We have utilized the Getz Lab CGA WES Characterization pipeline (https://github.com/broadinstitute/CGA_Production_Analysis_Pipeline) developed at the Broad Institute to call, filter and annotated somatic mutations and copy number variation. We modified this pipeline to call blood samples without matched controls. Hence, we employed the following tools: MuTect46, Strelka47, Orientation Bias Filter48, MAFPonFilter49, RealignmentFilter, ABSOLUTE50, GATK MuTect251, PicardTools51,52, Variant Effect Predictor53, and Oncotator54.
Usually the variant allele frequency (VAF) cutoff to call mutations is set at 0.02, below which it would be difficult to distinguish somatic mutations from contamination by other samples and sequencing artifacts. Thus, we aimed at estimating contamination to assure that the mutations are biologically relevant even when going below 0.02. We present a framework that allows one to include smaller clones in CHIP calling as a function of the single-sample contamination, providing greater resolution into the initial claim that CHIP is the presence of characteristic driver gene mutations in hematopoietic cells that occurs at a VAF of at least 0.02. This distinction of clonal mutations from contamination becomes especially important for those samples whose sample-to-sample contamination is in fact greater than 2% such that a mutation with a VAF of 0.02 would not be considered in our analysis if the sample’s contamination was about 3% (the range of contaminations in our samples was from roughly 1–8%). However, given our high depth of coverage (978×) we were able to confidently call mutations, distinct from sample-to-sample contamination and sequencing artifacts.
To estimate contamination of single samples, we used VerifyBamID55 using the ExAC56 VCF to test for germline SNPs with a minimum allele fraction of 0.25. In order to control for noise/artifacts with indel calling, we selected the youngest sample with no detectable CHIP mutations to use as an unmatched control for Strelka and MuTect2. Variants were classified as pathogenic driver mutations based on mutation type, position, and frequency in published reports8,19,57 and public databases58. The set of rules to consider a queried mutation as a “driver” mutation is outlined in supplementary table 2. The minimum number of alternate reads we chose to accept or reject to call a variant according to MuTect2 was not a hard cutoff but rather one determined as a function of the read depth, strand biases and contamination at a given site46. Consequently, the lowest number of accepted alternate reads was 4. Variants with allele fraction less than 0.01 were excluded and, except for DNMT3A, TET2, ASXL1, PPM1D, TP53, JAK2, SF3B1, SRSF2, mutations with VAF above 0.35 were also excluded since these often represent germline polymorphisms. To further confirm that our called mutations are somatic drivers, we excluded single nucleotide variant (SNV) mutations with a TLOD score below 6.3 (via MuTect2) and Insertion-Deletion (Indel) mutations with a QSI_NT score below 30 (via Strelka). As a consequence of bait preferences, some germline mutations can sometimes have putatively somatic allele fractions if they are in regions of poor map**. However, because Strelka and MuTect2 perform local realignment, these callers do not have deflated allele fractions in contrast to other callers that double count reference reads if they detect split reads, which is a consequence of structural variation. Finally, all variants were visually inspected in Integrated Genome Viewer (IGV)59.
We compared sequential samples taken from the same patient, in search for the presence of a mutation, called in one sample, in another sample from the same patient. To do that, we performed force-calling which is a technique that looks at the reads in a BAM file from a list of genomic coordinates and calculates the number of reads supporting an alternate allele at that location60.
Statistical analyses
OS was defined as the time from transplantation until death from any cause, with censoring at time last known to be alive. PFS was measured from ASCT to the date of disease progression or death from any cause, censoring at time last known to be alive and progression-free. Survival curves were estimated using the Kaplan Meier method, with variance and CIs estimated using Greenwood’s formula. Stratified Cox regression was used for time-to-event outcomes; hazard ratios (HR) and 95% confidence intervals (CI) were reported. Stratification was based on age (age groups: 20–29, 30–39, 40–49, 50–59, 60–69, 70–79), ISS of the disease61 and number of lines received prior to ASCT. p-Values were two-sided, and those <0.05 were considered statistically significant. All data were analyzed using R version 3.5.0 (R Core Team).
Death and occurrence of a TMN (namely, MDS and AML) were modeled as competing events. Wilcoxon rank-sum and Fisher’s exact tests were used for CHIP association with continuous and categorical variables, respectively. Ordinal variables with three or more groups were tested for association with CHIP using a Kruskal-Wallis test for singly-ordered contingency tables.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequencing data and basic phenotype data can be accessed via dbGaP accession code phs001323. All other remaining data are available within the article and supplementary files, or available from the authors upon request.
References
Kyle, R. A. & Rajkumar, S. V. Multiple myeloma. Blood 111, 2962–2972 (2008).
Attal, M. et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 366, 1782–1791 (2012).
McCarthy, P. L. et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 366, 1770–1781 (2012).
McCarthy, P. L. et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J. Clin. Oncol.35, 3279–3289 (2017).
Steensma, D. P. et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015).
**e, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Steensma D. P., et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015).
Gibson C. J., Steensma D. P. New insights from studies of clonal hematopoiesis. Clin. Cancer Res. 24, 4633–4642 (2018).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Zink, F. et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017).
Young, A. L., Challen, G. A., Birmann, B. M. & Druley, T. E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 7, 12484 (2016).
Abelson, S. et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 (2018).
Desai, P. et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 24, 1015–1023 (2018).
Young, A. L., Tong, R. S., Birmann, B. M. & Druley, T. E. Clonal hematopoiesis and risk of acute myeloid leukemia. Haematologica 104, 2410–2417 (2019).
Wong, T. N. et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518, 552–555 (2015).
Coombs, C. C. et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374–82 e4 (2017).
Gibson C. J., et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J. Clin. Oncol. 35, 1598-1605 (2017).
Takahashi, K. et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 18, 100–111 (2017).
Gillis, N. K. et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 18, 112–121 (2017).
Chitre, S. et al. Clonal hematopoiesis in patients with multiple myeloma undergoing autologous stem cell transplantation. Leukemia 32, 2020–2024 (2018).
Walker, B. A. et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 132, 587–597 (2018).
Walker, B. A. et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 33, 3911–3920 (2015).
Bolli, N. et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 5, 2997 (2014).
Maura, F. et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 10, 3835 (2019).
Lohr, J. G. et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 25, 91–101 (2014).
Wuilleme, S. et al. Assessment of tumoral plasma cells in apheresis products for autologous stem cell transplantation in multiple myeloma. Bone Marrow Transplant. 51, 1143–1145 (2016).
Kopp, H. G., Yildirim, S., Weisel, K. C., Kanz, L. & Vogel, W. Contamination of autologous peripheral blood progenitor cell grafts predicts overall survival after high-dose chemotherapy in multiple myeloma. J. Cancer Res Clin. Oncol. 135, 637–642 (2009).
Kahn, J. D. et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 132, 1095–1105 (2018).
Hsu, J. I. et al. PPM1D mutations drive clonal hematopoiesis in Rresponse to cytotoxic chemotherapy. Cell Stem Cell 23, 700–13 e6 (2018).
Boettcher, S. et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604 (2019).
Fuster, J. J. et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017).
Lode, L. et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica 103, e143–e6 (2018).
Jadersten, M. et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 29, 1971–1979 (2011).
Attal, M. et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N. Engl. J. Med. 376, 1311–1320 (2017).
Stadtmauer, E. A. et al. Autologous transplantation, consolidation, and maintenance therapy in multiple myeloma: results of the BMT CTN 0702 trial. J. Clin. Oncol. 37, 589–597 (2019).
Kumar, S. K. et al. Early versus delayed autologous transplantation after immunomodulatory agents-based induction therapy in patients with newly diagnosed multiple myeloma. Cancer 118, 1585–1592 (2012).
Dunavin, N. C. et al. Early versus delayed autologous stem cell transplant in patients receiving novel therapies for multiple myeloma. Leuk. Lymphoma 54, 1658–1664 (2013).
Gay, F. et al. Autologous transplant vs oral chemotherapy and lenalidomide in newly diagnosed young myeloma patients: a pooled analysis. Leukemia 31, 1727–1734 (2017).
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8, 1324 (2017).
Ha, G. et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 22, 1995–2007 (2012).
Ha, G. et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 24, 1881–1893 (2014).
Chapman, M. A. et al. Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472 (2011).
DePristo, M. A. et al. A framework for variation discovery and genoty** using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Ramos, A. H. et al. Oncotator: cancer variant annotation tool. Hum. Mutat. 36, E2423–E2429 (2015).
Jun, G. et al. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet 91, 839–848 (2012).
Karczewski, K. J. et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 45, D840–D845 (2017).
Lindsley, R. C. et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N. Engl. J. Med. 376, 536–547 (2017).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Stachler, M. D. et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 47, 1047–1055 (2015).
Greipp, P. R. et al. International staging system for multiple myeloma. J. Clin. Oncol. 23, 3412–3420 (2005).
Acknowledgements
We would like to thank Darlys Schott and Doreen Hearsey, as well as the Connell O’Reilly Cell Manipulation Facility and the Ted and Eileen Pasquarello Tissue Bank, for assistance in obtaining patient samples. We would also like to thank the International Myeloma Society (IMS) for their support. This work was supported by grants from the Multiple Myeloma Research Foundation (MMRF), Adelson Medical Research Foundation (AMRF), Stand Up to Cancer (SU2C) and the Leukemia and Lymphoma Society (LLS) awarded to Dr. Irene M. Ghobrial.
Author information
Authors and Affiliations
Contributions
Conceptualization, T.H.M., A.S.S., C.J.G., S. Manier, B.L.E., D.S.N., and I.M.G.; Methodology, J.P., R.R., M.L., R.S.P., C.L., S.H.A., B.R., E.M.V.A., J.J.K., C.S., S. Mehr, L.T., G.G., and D.S.N.; Investigation, T.H.M., A.H.N., M.C., D.H., M.B., S.T., K.H., H.D., M.M.I., and C.J.B.; Writing – Original Draft, T.H.M.; Writing – Review & Editing, T.H.M., A.S.S., C.J.G., D.A., R.L.S., N.M., K.C.A., D.P.S., J.P.L., P.G.R., J.R., B.L.E., R.J.S., L.T., D.S.N., and I.M.G.; Funding acquisition, I.M.G.; Resources, G.G., D.S.N., and I.M.G.; Supervision, D.S.N. and I.M.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: M.B.: Dava Oncology: Honoraria. N.C.M.: OncoPep: Other: Board of director. K.C.A.: Gilead: Membership on an entity’s Board of Directors or advisory committees; OncoPep: Equity Ownership, Scientific founder; Celgene: Consultancy; C4 Therapeutics: Equity Ownership, Scientific founder; Bristol Myers Squibb: Consultancy; Millennium Takeda: Consultancy. P.G.R.: Oncopeptides: Membership on an entity’s Board of Directors or advisory committees; BMS: Research Funding; Janssen: Membership on an entity’s Board of Directors or advisory committees; Karyopharm: Membership on an entity’s Board of Directors or advisory committees; Jazz Pharmaceuticals: Membership on an entity’s Board of Directors or advisory committees, Research Funding; Celgene: Membership on an entity’s Board of Directors or advisory committees, Research Funding; Amgen: Membership on an entity’s Board of Directors or advisory committees; Takeda: Membership on an entity’s Board of Directors or advisory committees, Research Funding. Amgen: Research Funding; Equillium: Research Funding; Kite/Gilead: Research Funding; Aleta Biotherapeutics: Consultancy; Avrobio: Consultancy; Celgene: Consultancy; Falcon Therapeutics: Consultancy; Talaris Therapeutics: Consultancy; TScan Therapuetics: Consultancy. B.L.E.: Celgene: Research funding; Deerfield: Research funding; GRAIL: Consultancy; Exo Therapeutics: scientific advisory boards; Skyhawk Therapeutics: scientific advisory boards. R.J.S.: Kiadis: Board of Directors; Juno: DSMB; Gilead: Consultancy; Neovii: Consultancy; Cugene: Consultancy; Jazz: Consultancy; Mana Therapeutics: Consultancy; VOR: Consultancy; Novartis: Consultancy. G.G.: receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig and POLYSOLVER. I.M.G.: Celgene: Consultancy; Janssen: Consultancy; BMS: Consultancy; Takeda: Consultancy; GSK: Consultancy; Sanofi: Consultancy; Karyopharm: Consultancy; Abbvie: Consultancy; Cellectar: Consultancy; Genentech: Consultancy; Adaptive: Consultancy. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mouhieddine, T.H., Sperling, A.S., Redd, R. et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun 11, 2996 (2020). https://doi.org/10.1038/s41467-020-16805-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16805-5
- Springer Nature Limited
This article is cited by
-
Bone marrow inflammation in haematological malignancies
Nature Reviews Immunology (2024)
-
Clonal hematopoiesis–derived therapy-related myeloid neoplasms after autologous hematopoietic stem cell transplant for lymphoid and non-lymphoid disorders
Leukemia (2024)
-
Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic malignancies
Signal Transduction and Targeted Therapy (2023)
-
Lymphoid clonal hematopoiesis: implications for malignancy, immunity, and treatment
Blood Cancer Journal (2023)
-
Clonal haematopoiesis - a novel entity that modifies pathological processes in elderly
Cell Death Discovery (2023)