

Abstract
The folate and methionine cycles are crucial for biosynthesis of lipids, nucleotides and proteins, and production of the methyl donor S-adenosylmethionine (SAM). 5,10-methylenetetrahydrofolate reductase (MTHFR) represents a key regulatory connection between these cycles, generating 5-methyltetrahydrofolate for initiation of the methionine cycle, and undergoing allosteric inhibition by its end product SAM. Our 2.5 Å resolution crystal structure of human MTHFR reveals a unique architecture, appending the well-conserved catalytic TIM-barrel to a eukaryote-only SAM-binding domain. The latter domain of novel fold provides the predominant interface for MTHFR homo-dimerization, positioning the N-terminal serine-rich phosphorylation region near the C-terminal SAM-binding domain. This explains how MTHFR phosphorylation, identified on 11 N-terminal residues (16 in total), increases sensitivity to SAM binding and inhibition. Finally, we demonstrate that the 25-amino-acid inter-domain linker enables conformational plasticity and propose it to be a key mediator of SAM regulation. Together, these results provide insight into the molecular regulation of MTHFR.
Similar content being viewed by others
Introduction
In humans, the folate and methionine cycles both generate products essential to cellular survival. Folate, the major cellular carrier of single carbon units, is required for the synthesis of purines and thymidine monophosphate. Within the methionine cycle, the methylation of homocysteine to methionine by methionine synthase (EC 2.1.1.13) produces an essential amino acid which may be used for protein synthesis or, crucially, be further converted to S-adenosylmethionine (SAM), a vitally important donor for the methylation of DNA, RNA and proteins as well as the creation of numerous methylated compounds. These two cycles intersect at the enzyme 5,10-methylenetetrahydrofolate reductase (MTHFR; E.C. 1.5.1.20). MTHFR catalyzes the physiologically irreversible reduction of 5,10-methylene-tetrahydrofolate (CH2-THF) to 5-methyl-tetrahydrofolate (CH3-THF), a reaction requiring FAD as a cofactor and NADPH as an electron donor. Since the product CH3-THF is exclusively used by methionine synthase, and only the demethylated form (THF) may be recycled back to the folate cycle, MTHFR commits THF-bound one-carbon units to the methionine cycle.
In accordance with this essential role, major and minor deficiencies of human MTHFR are the direct or indirect causes of human disease. Severe MTHFR deficiency (MIM #607093) is inherited in an autosomal recessive manner and is the most common inborn error of folate deficiency1 with ~200 patients known2. To date, over 100 different clinically relevant mutations in MTHFR have been described, the majority of which are of the missense type (n = 70, >60%) and private2. Milder enzyme deficiencies, due to single nucleotide polymorphisms of the MTHFR gene, have been associated with various common disorders. The most studied of these is p.Ala222Val (c.665C>T in NM_001330358, commonly annotated as c.677C>T), identified as a risk factor for an overwhelming number of multifactorial disorders, including vascular diseases, neurological diseases, various cancers, diabetes and pregnancy loss (see e.g. review by Liew and Gupta3).
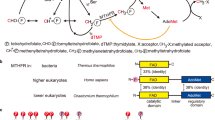
Human MTHFR is a 656 amino acid multi-domain protein (Fig. 1). The catalytic domain is conserved across evolution, and crystal structures of MTHFR from Escherichia (E.) coli4,5,6,7 and Thermus thermophilus8, in which the catalytic domain constitutes the entire sequence (Fig. 1), have been solved. These structures reveal the catalytic domain to form a β8α8 (TIM) barrel and have uncovered residues critical for binding the cofactor FAD4, the electron donor NADPH (NADH in bacteria7) and the product CH3-THF5,6,7. The bacterial structures, together with activity assay of trypsin cleaved porcine MTHFR9, indicate that the catalytic domain is sufficient for the entire catalytic cycle. Eukaryotic MTHFR orthologs additionally possess a C-terminal regulatory domain that is connected to the catalytic domain by a linker sequence (Fig. 1). This C-terminal domain is able to bind SAM, resulting in allosteric inhibition of enzymatic activity10, an effect which is very slow11 and can be reversed by binding to S-adenosylhomocysteine (SAH)12,13, the demethylated form of SAM.

Schematic representation of MTHFR. Domain organization of MTHFR orthologs across evolution. Numbers given represent approximate amino acid boundaries in human MTHFR corresponding to NP_005948. In brackets is shown representative species within each category
Human MTHFR further contains a 35 amino acid serine-rich region at the very N-terminus which is not found in MTHFR orthologs of bacteria, yeast or even lower animals (Fig. 1). This region has been identified to be multiply phosphorylated following heterologous expression in insect cells14 and yeast15, or following immunoprecipitation from human cancer cell lines14,15, suggesting Thr34 is the priming position. The cellular relevance of this modification remains unclear, although one group has suggested that phosphorylation at Thr34 can be accomplished by CDK1/cyclin B1 (ref. 2a). All phosphorylation sites were considered to have partial occupancy, since no residues were phosphorylated in every tryptic peptide analysed (Supplementary Fig. 1). Of these, 11 phosphorylated amino acids (Ser9, Ser10, Ser18, Ser20, Ser21, Ser23, Ser25, Ser26, Ser29, Ser30, Thr34) were within the N-terminal serine-rich region, including the putative phosphorylation determining residue Thr34 (Fig. 2a). Additionally, we found phosphorylation of three further amino acids in the catalytic domain (Tyr90, Thr94, Ser103) and two in the regulatory domain (Ser394, Thr451). Up to ten phosphorylation sites were identified to be occupied simultaneously, whereby treatment with calf intestine alkaline phosphatase (CIP) resulted in removal of 9 (Fig. 2b) or 10 (Fig. 2c) phosphate groups, as identified by denaturing and native mass spectrometry, respectively. To examine the importance of the N-terminal serine-rich region to global protein phosphorylation, we produced recombinant HsMTHFR38–644, which removes the N-terminal 37 amino acids, including the entire serine-rich region (Fig. 1) as well as the poorly conserved C-terminal 12 amino acids predicted to be of high disorder (Supplementary Fig. 2). As purified HsMTHFR38–644 was not found to be phosphorylated by phosphorylation map** (Supplementary Fig. 3a), or native mass spectrometry (Supplementary Fig. 3b), and treatment with CIP did not alter the protein molecular mass (Fig. 2d). Therefore, the primary determinant of HsMTHFR phosphorylation resides within the N-terminus.

Phosphorylation status of HsMTHFR1–656 and HsMTHFR38–644. a Phosphorylation map** of HsMTHFR1–656. The protein sequence is given as amino acids in single letter code, including the C-terminal His/flag-tag (underlined). Black font represents amino acids identified by the mass spectrometer (covered), blue font represents amino acids not identified (non-covered), red font represents phosphorylated amino acids. Domains are coloured as in Fig. 1. b Dephosphorylation of HsMTHFR1–656 following treatment with CIP. Treatment time at 37 °C is given. Large number above peaks represents number of phosphate groups attached. Proteins were analysed by denaturing mass spectrometry. amu, atomic mass units. c Native mass spectrometry analysis of HsMTHFR1–656 before and after treatment with CIP. Upper panel: as purified (untreated) protein. Monomer represents protein bound to 1 FAD plus 10 phosphate groups (expected mass: 76831.16 amu); dimer represents protein bound to 2 FADs and 1 SAM plus 20 phosphate groups (expected mass: 154060.74 amu). Lower panel: protein following 180 min treatment with CIP. Monomer represents protein bound to 1 FAD (expected mass: 76031.16 amu); dimer represents protein bound to 2 FADs and 1 SAH (expected mass: 152446.74 amu). Expected sizes: protein without first methionine, 75245.6 amu; FAD, 785.56 amu; SAM, 398.44 amu; SAH, 384.42 amu, phosphate, 80.00 amu. * indicates a truncated protein representing amino acids 353–663 (expected mass: 36136.6 amu). d HsMTHFR38–644 before and after treatment with CIP. Treatment time at 37 °C is given. Protein was analysed by denaturing mass spectrometry. e Native mass spectrometry of HsMTHFR1–656 identifying sequential binding of SAM or SAH. Graphs represent areas zoomed in on dimeric protein. Upper panel: As purified (untreated) protein. Middle panel: control protein (heated in assay buffer for 180 min without CIP). Bottom panel: treated protein (heated in assay buffer for 180 min with CIP). Expected size of protein with 2 FAD bound and 20 phosphates: 153662.3. Expected size of protein with 2 FAD bound and 0 phosphates: 152062.32. Expected size of SAM: 398.44, SAH: 384.41
Phosphorylation does not alter MTHFR kinetic parameters
Phosphorylation has been described to alter MTHFR kinetics, resulting in moderately decreased catalytic activity as measured by the NADPH-menadione oxidoreductase assay14,7). Twice as many of the mutation sites are found in the catalytic domain (n = 38) as the regulatory domain (20), with the remainder (6) found in the linker region. By proportion, however, the linker region has a higher density (24% of the sequence) of mutation sites than the catalytic (11%) and regulatory (7%) domains. Additionally, a number of sites in the catalytic and regulatory domains are in direct contact with the linker region. Further, the most severe mutations, those found either homozygously or in conjunction with a truncating mutation to result in enzymatic activity below 1.5% of control activity in patient fibroblasts20, cluster in the catalytic domain and the first two aa of the linker region, most of which are located where the linker meets the catalytic domain (Supplementary Fig. 7). Together, this analysis underscores the importance of the linker region to proper protein function.
An asymmetric MTHFR dimer with inter-domain flexibility
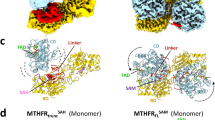
The HsMTHFR38–644 structure reveals a homodimer (Fig. 4b), consistent with native mass spectrometry (Supplementary Fig. 3b) and previous investigation of mammalian MTHFR by size exclusion chromatography and scanning transmission electron microscopy9. It was previously thought that MTHFR homodimerizes in a head-to-tail manner, where the regulatory domain of one subunit interacts with the catalytic domain of the other subunit13. Unexpectedly, in our structure dimerization is mediated almost entirely by the regulatory domain (Fig. 4b), although the first ordered residue in chain A (Glu40) is located around 5–6 Å from the regulatory domain of chain B (e.g. Glu553, Arg567). The N-terminal sequence that is either not present (Ser-rich phosphorylation region, aa 1–37) or present but disordered (aa 38–39) in the HsMTHFR38–644 structure will likely project towards the interface of the two regulatory domains (Fig. 4c), and may contribute further to the dimer contacts.
The essential interfacial residues from the regulatory domain are contributed predominantly from the two central β-sheets, including a β-turn (β11–β12), strand β16, and the loop encompassing Asn386–Asn391 (Supplementary Fig. 8), which buries in total ~1330 Å2 of accessible surface. Half of the sites of missense mutations in the regulatory domain causing MTHFR deficiency (n = 10, Supplementary Fig. 7) either participate in, or are within two residues of, the dimerization site.
Within the homodimer, each of the two catalytic domains is presented away from the dimeric interface and their active sites are at opposite ends of the overall shape and face away from each other (Fig. 4b). In this arrangement, the catalytic domain is not involved in oligomerization, unlike bacterial and archaeal MTHFR proteins (Supplementary Fig. 9). This said, the N-terminus of the HsMTHFR38–644 construct is projecting towards the dimer interface. A direct consequence of the dimeric architecture is that the HsMTHFR catalytic domain displays a large degree of flexibility in relative orientation with the regulatory domain. In fact, this is reflected in our structure whereby the catalytic domain of one dimer subunit (chain A) is ordered, while that of the other dimer subunit (chain B) is highly disordered, to the extent that only main chain atoms of the amino acid 40–58, 129–134 and 155–342 in chain B could be modelled.
Dynamics of MTHFR observed by solution scattering
Our HsMTHFR38–644 crystal structure has captured the snapshot of an asymmetric dimer whereby the two catalytic domains have different orientations with respect to their own regulatory domains (Supplementary Fig. 10). We applied small-angle X-ray scattering (SAXS) to understand better the different conformational variations assumed by the protein in solution. Superimposition of the theoretical scattering curve back-calculated from the crystal structure dimer against experimental data obtained from HsMTHFR38–644 in solution revealed a poor fit (Chi2 14.8; Fig. 5), suggesting this is not the predominant conformation in solution. However, by employing CORAL21 to simulate relaxation of the relative orientations of the catalytic and regulatory domains (by allowing flexibility in residues 338–345 of the linker), and thus also permitting rigid body movement of these subunits in relative orientation to each other, we obtained a significantly improved fit (Chi2 5.5; Fig. 5). Thus, consistent with our finding from the crystal structure, HsMTHFR retains a significant degree of intra- and inter-domain conformational flexibility in solution.

SAXS analysis of HsMTHFR38–644 and HsMTHFR1–656 phosphorylated and dephosphorylated. SAXS analysis of HsMTHFR38–644. Experimental scattering profile is shown in black, theoretical scattering curve of the HsMTHFR38–644 dimer observed in the crystal is in green and that of the rigid body modelling by CORAL is in red. Chi2 was determined by CRYSOL53
To further investigate the influence of phosphorylation on protein conformation, we next collected SAXS data for full-length HsMTHFR1–656 as purified (i.e. phosphorylated and bound with SAM) and treated with CIP (i.e. dephosphorylated and bound with SAH). The experimental scattering curves for as purified and CIP-treated HsMTHFR1–656 gave rise to slightly different profiles and derived parameters (Supplementary Fig. 11, Supplementary Table 1), although both protein forms are consistent with a dimeric configuration. These data were further corroborated by charge radius analysis of native phosphorylated and dephosphorylated HsMTHFR1–656 by electrospray ionization mass spectrometry, showing that the charge-distribution of protein ions is shifted between the two protein forms. This may indicate a conformational change equivalent to a 0.5% change in radius (Supplementary Fig. 12). Together, we interpret these results to suggest that the phosphorylated SAM-bound form of the protein may present a different conformation to the dephosphorylated SAH-bound form, which merits future investigation using alternative methods.
Subtle features provide for eukaryotic NADPH specificity
The MTHFR catalytic domain adopts a TIM-barrel structure evolutionarily conserved across all kingdoms. In addition to HsMTHFR38–644, we further determined the catalytic domain structure of the yeast homologue MET12 (ScMET121–301) to 1.56 Å resolution (Table 2). This enables a structural comparison across mammalian (HsMTHFR), low eukaryotic (ScMET12) and bacterial (E. coli, H. influenzae, T. thermophilus) orthologues. Consistent with their sequence conservation (Supplementary Fig. 13), the catalytic domains have highly superimposable folds (main chain RMSD: 1.85 Å), although distinct local differences are found in low homology loop regions (Fig. 6a, 1–2) and helices (Fig. 6a, 3–4). Additionally, the first helix of the catalytic domain (α1) is observed in different orientations among these structures (Supplementary Fig. 14). There is sequence divergence of helix α1 among prokaryotes, lower and higher eukaryotes (Supplementary Fig. 15). In HsMTHFR (which contains, in its biological sequence but not present in the crystallized construct, the serine-rich phosphorylation region N-terminal to the catalytic domain), ScMET12 and T. thermophilus MTHFR, this helix α1 is projected towards the interface between catalytic and regulator domains.

Structural examination of the HsMTHFR38–644 catalytic domain. a Structural alignment of HsMTHFR38–644 (cyan) with EcMTHFR (grey) and ScMET121–301. Four sites of important differences are indicated by arrows (1–4). α-helices of HsMTHFR38–644 are indicated for orientation. b Binding pocket of FAD. FAD is shown in green sticks, residues contributing to FAD binding are labelled and shown in black sticks. c Binding pocket of NAD(P)H. NADH is taken from an overlay of EcMTHFR (PDB: IZRQ) with HsMTHFR38–644 but for clarity EcMTHFR is not shown. FAD is shown in green sticks, NADH in brown sticks, residues expected to contribute to NADH binding are labelled and shown in black sticks. d Binding pocket of CH3–THF. CH3–THF is taken from an overlay of EcMTHFR (PDB: 2FMN) with HsMTHFR38–644 but for clarity EcMTHFR is not shown. FAD is shown in green sticks, CH3-THF in yellow sticks, residues expected to contribute to CH3-THF binding are labelled and shown in black sticks
In HsMTHFR38–644, clear electron density for FAD was observed in the TIM barrel of chain A (Supplementary Fig. 5a). However, there is high disorder in the TIM-barrel of chain B particularly around the FAD binding site, implying a low ligand occupancy of the ligand, although native mass spectrometry of the crystallized construct indicated two FADs bound per homodimer (Supplementary Fig. 3b). Analysis of the FAD binding residues in HsMTHFR chain A (Fig. 6b) reveals perfect overlap with those predicted from the EcMTHFR structure4. These include Thr129, Arg157, Ala175 and Ala195 of HsMTHFR, which were associated with in vitro FAD responsiveness when mutated in severe MTHFR deficiency20,22 (Fig. 6b; Supplementary Fig. 7).
The bi bi kinetic mechanism of MTHFR necessitates the electron donor NAD(P)H and substrate CH2-THF to interact in turn with FAD for transfer of the reducing equivalents, and hence to share the same binding site. In our structures, the FAD ligand adopts a conformation poised to expose the si face of the isoalloxazine ring for the incoming NAPDH and CH2-THF. However, instead of trap** the electron donor or substrate (despite multiple attempts at co-crystallization), the binding site in ScMET121–301 and subunit A of HsMTHFR38–644 is blocked by a crystal packing interaction from a nearby symmetry mate, making π–π stacking interactions with the FAD ligand (Supplementary Fig. 16). By contrast, no crystal packing interaction is found in the chain B binding site of HsMTHFR38–644, explaining the overall mobility and disorder of its catalytic domain.
Superimposing the HsMTHFR38–644 structure with that of EcMTHFR bound with NADH (Fig. 6c) and CH3-THF (Fig. 6d) demonstrates that the human enzyme has largely preserved the same shared binding site found in prokaryotes, with Gln228, Gln267, Lys270, Leu271 and Leu323 likely to be important for interacting with both NAD(P)H and CH3-THF. EcMTHFR preferentially utilizes NADH23, and its NADH-bound structure reveals a highly uncommon bent conformation24 for the electron donor, where the nicotinamide ring stacked over the adenine base mediates π–π interactions7. Our activity assay of HsMTHFR38–644 and HsMTHFR1–656 clearly demonstrates an ~100-fold preference for NADPH compared to NADH as an electron donor (Table 1), in agreement with previous enzyme studies from pig11,25 and rat11 MTHFRs.
Within the HsMTHFR active site, we did not identify any obvious differentiating features surrounding the modelled NADH, which could indicate how the extra 2′-monophosphate group on the NADPH ribose is accommodated (Supplementary Fig. 17). It is also unclear if HsMTHFR actually binds NADPH in a similar manner as NADH for EcMTHFR, considering there is only one report in the literature documenting a compact stacked conformation for NADPH26. Modelling an NADPH ligand with such a stacked conformation onto the HsMTHFR38–644 structure reveals severe steric clashes with helix α8 (Supplementary Fig. 17), which creates the floor of the NAD(P)H binding site (e.g. via Gln267, Lys270 and Leu271). Helix α8 is poorly aligned with bacterial and low eukaryotic orthologues in both amino acid sequence (Supplementary Fig. 13) and structural topology (Fig. 6a). The equivalent helix in EcMTHFR harbours the residue Phe223, which is crucial to NADH binding7 and moves to accommodate substrate release5. Notably, this residue is not conserved in HsMTHFR and ScMET12, replaced by Gln267 and Ala229, respectively. (Supplementary Fig. 13). Therefore, given its position and mobility, we propose that residue(s) on helix α8 in HsMTHFR may play a role in the specificity for NADPH and likely also substrate binding/release.
A novel fold for the SAM-binding regulatory domain
The HsMTHFR38–644 structure provides a view of the 3D arrangement of the regulatory domain unique to eukaryotic MTHFR. The core of this fold comprises two mixed β-sheets of five strands each (β9↑-β17↑-β16↓-β12↑-β11↓ and β10↓-β13↑-β18↓-β14↑-β15↓) (Supplementary Fig. 6). Strand β10 from one sheet forms a continuous segment with β11 from the other sheet, and similarly β12 from one β-sheet continues onto β13 of the other sheet. The threading of the two central β-sheets are interspersed with three loop extensions containing different numbers of α-helices (α12–α15, α16, and α17–α18). To the best of our knowledge, the MTHFR regulatory domain represents a unique SAM binding architecture distinct from the 18 known classes of SAM-dependent methyltransferases and non-methyltransferases27 (Supplementary Fig. 18). Further, a DALI search of this domain28 did not yield any structural homologue, and we found no existing annotation in PFAM/CATH/SCOP databases and no sequence for this domain beyond eukaryotic MTHFR homologues. Therefore, this appears to be a novel fold utilized only by MTHFR for SAM binding/inhibition.
In our structure, SAH is bound in an extended conformation within the part of the regulatory domain (Fig. 7a) that faces the catalytic domain. Indeed, part of the binding site is constituted by the linker region itself. The ligand is sandwiched between the loop segment preceding α15 (N456DEPLAAET464) and the first strand β10 (T481INSQ485) of the central β-sheets, where a number of conserved residues are found. For example, Thr481 (conserved in 96% of 150 orthologues; Consurf29) and Ser484 (98%) hydrogen-bond to the SAH adenine moiety, while Glu463 (99%) and Thr464 (62%) fixate the ribose hydroxyl groups. The strongest sequence conservation in the SAH binding site is found around the homocysteine moiety, including Pro348 (invariant) and Trp349 (99%) from the linker region, as well as Thr560 and Thr573 (both invariant) at the start and end of the β15-β16 turn. The SAH homocysteine sulphur atom is loosely contacted by Glu463 (3.8 Å) and Ala368 (3.7 Å). SAM is expected to bind to the same site in the regulatory domain, in a similar extended configuration as SAH and requiring the same set of binding residues. However, the additional methyl group in the sulphonium centre of SAM would create a steric clash to the Ala368 position of the structure (inter-residue distance ~2.0 Å between heteroatoms, and <1.5 Å between hydrogen atoms) (Supplementary Fig. 19). Although not strictly conserved (45% of 150 orthologues), conservation of Ala368 follows a similar evolutionary pattern as the MTHFR domain organization (Fig. 1): in higher animals alanine is invariant; lower animals may accommodate a serine; while lower eukaryotes often incorporate a bulky residue (e.g. lysine) (see Supplementary Fig. 15). Therefore, in higher organisms such as humans, SAM binding likely results in conformational rearrangement of the loop region containing Ala368 to accommodate its methyl moiety.

SAH/SAM binding and conformational change. a The SAH binding site. Amino acids that contribute to binding are labelled and shown in black sticks. SAH is shown in green sticks. b Size exclusion chromatography of HsMTHFR with various N-terminal truncations following incubation with SAM (dashed lines), SAH (dotted lines), or buffer (apo; solid line). For each N-terminally truncated construct, the corresponding structure is shown. c Size exclusion chromatography of HsMTHFR348–656 proteins without (wt) or with (wt-SAM) pre-incubation with SAM. Mutated HsMTHFR348–656 proteins were pre-incubated with SAM
The linker mediates SAM-dependent conformational change
Since there is no direct interface between the active site of the catalytic domain and the regulatory domain (Fig. 4), SAM binding must elicit enzymatic inhibition via a conformational change propagated from the regulatory to catalytic domain. The most likely effector of this conformational change is the extended linker region (defined as aa 338–362), since it makes multiple contacts to both the regulatory and catalytic domains (Fig. 4) and forms part of the SAM/SAH binding site (Fig. 7a). To investigate the potential of this region to elicit conformational change following SAM binding, we generated recombinant HsMTHFR proteins consisting of the regulatory domain alone attached to progressively shorter linker regions, where the N-terminus of these constructs would become Pro348 (HsMTHFR348–656), Arg357 (HsMTHFR357–656) and Arg377 (HsMTHFR377–656) (Fig. 7b; Supplementary Fig. 2). All three constructs are sufficient to bind SAM and SAH, as demonstrated by dose-dependent increases in thermostability by differential scanning fluorimetry when exposed to increasing concentrations of each ligand (Supplementary Fig. 20a). This again reinforces the catalytic and regulatory domains as separate binding modules for their cognate ligands (FAD/NADPH/CH3-THF vs SAM/SAH respectively).
We employed analytical size exclusion chromatography (aSEC) as a means to study solution behaviour of the MTHFR regulatory domain in response to SAM/SAH binding. Exposure of MTHFR348–656 to either SAH or SAM resulted in shifts of elution volume (Ve) compared to as purified (apo-) protein (Fig. 7b), in contrast to HsMTHFR1–656 and HsMTHFR38–644, which did not show changes in Ve despite SAH or SAM binding (Supplementary Fig. 21). Importantly, for MTHFR348–656, SAM resulted in a leftward Ve shift (suggestive of a larger hydrodynamic volume) and SAH a rightward shift (suggestive of a smaller hydrodynamic volume) (Fig. 7b). By contrast, MTHFR357–656 showed a shift in Ve only when exposed to SAM, and MTHFR377–656 did not change when exposed to either ligand (Fig. 7b). A similar pattern of results were observed when using purified recombinant mouse MTHFR of the same protein boundaries (Supplementary Fig. 20a and b). Therefore, we conclude that residues within 357–377 must contribute to change of protein state upon SAM binding, which we interpret as a change in conformation.
Next we carried out site-directed mutagenesis to define residues involved in SAM binding, and/or SAM-mediated conformational change as observed in the aSEC experiment. We reasoned that mutation of Glu463 (which hydrogen-bonds a ribose oxygen) could lead to loss of SAH/SAM binding, and thus conformational change. Indeed, conservative mutation of Glu463 to either aspartate (p.E463D) or glutamine (p.E463Q) on MTHFR348–656 resulted in protein that could no longer bind SAM (Supplementary Fig. 20c), nor change conformation in its presence (Fig. 7c). We further hypothesized that mutation of Ala368 (in close proximity to the SAM/SAH sulphonium centre) to a smaller residue (glycine: p.A368G) may not have an effect on binding or conformational change, while mutation to a larger residue (leucine: p.A368L) might reduce the ability of the linker region to sense SAM binding. Correspondingly, p.A368L resulted in protein which retained the ability to bind SAM, but was less sensitive to change in its presence, while p.A368G did not change either of these properties. (Fig. 7c, Supplementary Fig. 20c). These experiments conclusively pinpoint Glu463 as crucial to SAM binding and Ala368 to SAM sensing, representing a mechanism that could transmit a ligand-bound signal from regulatory to catalytic domain of the protein.
Discussion
Catalytic regulation by phosphorylation and SAM binding distinguishes human MTHFR from its bacterial (which do not have phosphorylation or SAM binding regions) and lower eukaryotic (which do not have a phosphorylation region) counterparts. Until now, the molecular basis of how these two allosteric events modulate the catalytic machinery was entirely unknown, due to the absence of a structural context. Now, our structure-guided study has provided two major discoveries in this area: (1) identification of an extensive linker region that functionally connects SAM-binding in the regulatory domain with inhibition in the catalytic domain and (2) demonstration of the concerted effects of phosphorylation and SAM binding, individually mediated by regions more than 300 amino acids apart (Supplementary Fig. 22).
We mapped the entire phosphorylation landscape of HsMTHFR, revealing phosphorylated Ser/Thr not only at the far N-terminus (n = 11) as predicted from the sequence but also within the catalytic (3) and regulatory (2) domains. Many of the N-terminal phosphorylation sites identified are consistent with previous mutation analysis14, including Thr34 (refs. 14,15,Denaturing intact mass analysis Reversed-phase chromatography was performed in-line prior to mass spectrometry using an Agilent 1290 uHPLC system. Concentrated protein samples were diluted to 0.02 mg mL−1 in 0.1% formic acid and 50 µL was injected on to a 2.1 mm × 12.5 mm Agilent Zorbax 5um 300SB-C3 guard column housed in a column oven set at 40 °C. The solvent system used consisted of 0.1% formic acid in LC-MS grade water (solvent A) and 0.1% formic acid in LC-MS grade methanol (solvent B). Chromatography was performed as follows: initial conditions were 90% A and 10% B and a flow rate of 1.0 mL min−1. A linear gradient from 10% B to 80% B was applied over 35 s. Elution then proceeded isocratically at 95% B for 40 s followed by equilibration at initial conditions for a further 15 s. Protein intact mass was determined using a 6530 QTOF mass spectrometer (Agilent). The instrument was configured with the standard ESI source and operated in positive ion mode. The ion source was operated with the capillary voltage at 4000 V, nebulizer pressure at 60 psig, drying gas at 350 °C and drying gas flow rate at 12 L min−1. The instrument ion optic voltages were as follows: fragmentor 250 V, skimmer 60 V and octopole RF 250 V. Mass spectrometry of MTHFR under native conditions was performed on the same MS instrument, and is described in detail elsewhere51. Briefly, 50 µg of protein was desalted and exchanged into 50 mM ammonium acetate pH 6.5 using three rounds of size exclusion spin column purification ("Micro BioSpin 6", Biorad) following the manufacturer’s instructions. Approximately 50 µL was infused directly into the mass spectrometer via syringe pump at a rate of 6 µL min−1. The instrument was fitted with a standard source using a nebulizer pressure of 17 psi, drying gas flow rate 5 L min−1 and drying gas temperature 325 °C. The instrument was operated in positive ion, 1 GHz detector mode with fragmentor voltage 430 V, nebulizer pressure set to 17 psi, and collision gas pressure and energy at 0. Spectra were deconvoluted using the MaxEnt functionality within the Masshunter software package (Agilent) and multimeric states were determined manually using a charge table. The radius for native protein charge states was determined using the calculation z = R1.5 or R = z × 101/1.5 where z is the charge state and R is the radius in Ångstroms if the protein is assumed to be a sphere52. To allow comparison between spectra, ion intensities were normalized by dividing each value by the largest in that spectrum. Normalized ion intensity was plotted against charge radius for each ion. Between 20 and 100 µg MTHFR was reduced in 100 µL of 100 mM ammonium bicarbonate buffer, pH 7.5 by addition of 1 µL of 1 M DTT and incubation at 56 °C for 40 min. Alkylation was performed by addition of 4 µL of saturated iodoacetamide solution and incubated at room temperature in the dark for 20 min. Endoprotease digestion was performed using either trypsin, Smart Digest trypsin (Thermo) or pepsin. Trypsin in 100 mM ammonium bicarbonate buffer, pH 7.5 was added in the ratio 20:1 w/w and incubated at 37 °C overnight. Smart Digest trypsin was incubated at 70 °C for 1 h following the manufacturer’s instructions. Pepsin digestion was performed in 0.1 M HCl in the ratio 20:1 w/w and incubated at 37 °C overnight. LC-MSMS analyses were performed using both whole endoprotease digests and metal oxide-affinity enriched samples. Metal oxide-affinity enrichment for phosphopeptides was performed using home made spin columns containing a mixed bed of 2.5 mg Titansphere TiO2 chromatography matrix (GL Sciences) and 2.5 mg ZrO2 powder. Non-phospopeptides were eluted using 80% ACN, 300 mg mL−1 DHB, 0.1% TFA. Phosphopeptides were eluted in 25% ammonium hydroxide, 40% ACN. Samples were dried-down by rotary evaporation and re-suspended in 5 µL of 2% ACN, 0.1% FA prior to LC-MSMS. Analysis was performed using a Dionex U3000 nanoHPLC coupled to a Bruker Esquire HCT ion trap mass spectrometer. Peptides were separated using a 200 µm × 5 cm Pepswift PS-DVB monolithic column (Thermo, USA). A gradient was developed from 2 to 17% B over 4 min, then 17–42% B over 2 min. The column was washed at 92% B for 1 min and finally equilibrated at 2% B for 6 min at a flow rate of 2.5 µL min−1. Buffer A was 0.1% formic acid in LC-MS grade water: Buffer B was 0.1% formic acid, 80% LC-MS grade ACN. MSMS was performed in data-dependent mode with a scan rate of 26,000 m z−1 s−1 with three precursors per MS1 scan and active exclusion for 20 s. Charge state selection was +1, +2 and +3. Automated data analysis was performed using Data Analysis v 4.0 (Bruker). Database searching was performed using an in-house Mascot server, in which the MTHFR database was searched for variable modifications including oxidation (M) and phosphorylation (ST or Y) with an MS tolerance of 1.4 Da, MSMS tolerance of 0.5 Da, partials at 4, C13 at 1 and charge states of +1, +2, +3. All putative phosphopeptide assignments were evaluated manually with an assumed false discovery rate of zero. Composite MSMS for MTHFR coverage was 92%. The crystal structures of HsMTHFR and ScMET12 have been deposited in the protein data bank (PDB) with the identifiers 6CFX and 6FNU, respectively. The full mass spectrometry phosphorylation dataset has been submitted to MassIVE with the accession number MSV000082179. All other data are available from the corresponding authors upon reasonable request.Native mass spectrometry
Phosphorylation map**
Data availability
References
Watkins, D. & Rosenblatt, D. S. in The Online Metabolic and Molecular Bases of Inherited Disease (eds Valle, D. et al.) Ch. 155 (McGraw-Hill Medical, New York City, 2017).
Froese, D. S. et al. Mutation update and review of severe methylenetetrahydrofolate reductase deficiency. Hum. Mutat. 37, 427–438 (2016).
Liew, S. C. & Gupta, E. D. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: epidemiology, metabolism and the associated diseases. Eur. J. Med. Genet. 58, 1–10 (2015).
Guenther, B. D. et al. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat. Struct. Biol. 6, 359–365 (1999).
Lee, M. N. et al. Functional role for the conformationally mobile phenylalanine 223 in the reaction of methylenetetrahydrofolate reductase from Escherichia coli. Biochemistry 48, 7673–7685 (2009).
Pejchal, R. et al. Structural perturbations in the Ala--Val polymorphism of methylenetetrahydrofolate reductase: how binding of folates may protect against inactivation. Biochemistry 45, 4808–4818 (2006).
Pejchal, R., Sargeant, R. & Ludwig, M. L. Structures of NADH and CH3-H4 folate complexes of Escherichia coli methylenetetrahydrofolate reductase reveal a spartan strategy for a **-pong reaction. Biochemistry 44, 11447–11457 (2005).
Igari, S. et al. Properties and crystal structure of methylenetetrahydrofolate reductase from Thermus thermophilus HB8. PLoS ONE 6, e23716 (2011).
Matthews, R. G., Vanoni, M. A., Hainfeld, J. F. & Wall, J. Methylenetetrahydrofolate reductase. Evidence for spatially distinct subunit domains obtained by scanning transmission electron microscopy and limited proteolysis. J. Biol. Chem. 259, 11647–11650 (1984).
Sumner, J., Jencks, D. A., Khani, S. & Matthews, R. G. Photoaffinity labeling of methylenetetrahydrofolate reductase with 8-azido-S-adenosylmethionine. J. Biol. Chem. 261, 7697–7700 (1986).
Kutzbach, C. & Stokstad, E. L. Mammalian methylenetetrahydrofolate reductase. Partial purification, properties, and inhibition by S-adenosylmethionine. Biochim. Biophys. Acta 250, 459–477 (1971).
Daubner, S. C. & Matthews, R. G. Purification and properties of methylenetetrahydrofolate reductase from pig liver. J. Biol. Chem. 257, 140–145 (1982).
Yamada, K., Chen, Z., Rozen, R. & Matthews, R. G. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc. Natl. Acad. Sci. USA 98, 14853–14858 (2001).
Yamada, K., Strahler, J. R., Andrews, P. C. & Matthews, R. G. Regulation of human methylenetetrahydrofolate reductase by phosphorylation. Proc. Natl. Acad. Sci. USA 102, 10454–10459 (2005).
Marini, N. J. et al. The prevalence of folate-remedial MTHFR enzyme variants in humans. Proc. Natl. Acad. Sci. USA 105, 8055–8060 (2008).
Zhu, B., **ahou, Z., Zhao, H., Peng, B. & Xu, X. MTHFR promotes heterochromatin maintenance. Biochem. Biophys. Res. Commun. 447, 702–706 (2014).
Li, X. et al. Polo-like kinase 1 (PLK1)-dependent phosphorylation of methylenetetrahydrofolate reductase (MTHFR) regulates replication via histone methylation. Cell Cycle 16, 1933–1942 (2017).
Suormala, T., Gamse, G. & Fowler, B. 5,10-Methylenetetrahydrofolate reductase (MTHFR) assay in the forward direction: residual activity in MTHFR deficiency. Clin. Chem. 48, 835–843 (2002).
Matthews, R. G. Methylenetetrahydrofolate reductase from pig liver. Methods Enzymol. 122, 372–381 (1986).
Burda, P. et al. Insights into severe 5,10-methylenetetrahydrofolate reductase deficiency: molecular genetic and enzymatic characterization of 76 patients. Hum. Mutat. 36, 611–621 (2015).
Petoukhov, M. V. et al. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 (2012).
Burda, P. et al. Functional characterization of missense mutations in severe methylenetetrahydrofolate reductase deficiency using a human expression system. J. Inherit. Metab. Dis. 40, 297–306 (2017).
Sheppard, C. A., Trimmer, E. E. & Matthews, R. G. Purification and properties of NADH-dependent 5, 10-methylenetetrahydrofolate reductase (MetF) from Escherichia coli. J. Bacteriol. 181, 718–725 (1999).
van den Heuvel, R. H. et al. Structural studies on flavin reductase PheA2 reveal binding of NAD in an unusual folded conformation and support novel mechanism of action. J. Biol. Chem. 279, 12860–12867 (2004).
Matthews, R. G. & Kaufman, S. Characterization of the dihydropterin reductase activity of pig liver methylenetetrahydrofolate reductase. J. Biol. Chem. 255, 6014–6017 (1980).
Bhattacharyya, S., Dutta, A., Dutta, D., Ghosh, A. K. & Das, A. K. Structural elucidation of the NADP(H) phosphatase activity of staphylococcal dual-specific IMPase/NADP(H) phosphatase. Acta Crystallogr. D Struct. Biol. 72, 281–290 (2016).
Gana, R., Rao, S., Huang, H., Wu, C. & Vasudevan, S. Structural and functional studies of S-adenosyl-L-methionine binding proteins: a ligand-centric approach. BMC Struct. Biol. 13, 6 (2013).
Holm, L. & Laakso, L. M. Dali server update. Nucleic Acids Res. 44, W351–W355 (2016).
Ashkenazy, H., Erez, E., Martz, E., Pupko, T. & Ben-Tal, N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 (2010).
Wang, J. X., Lee, E. R., Morales, D. R., Lim, J. & Breaker, R. R. Riboswitches that sense S-adenosylhomocysteine and activate genes involved in coenzyme recycling. Mol. Cell 29, 691–702 (2008).
Zheng, W. et al. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc. 134, 18004–18014 (2012).
Wise, C. K., Cooney, C. A., Ali, S. F. & Poirier, L. A. Measuring S-adenosylmethionine in whole blood, red blood cells and cultured cells using a fast preparation method and high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 696, 145–152 (1997).
Loehrer, F. M. et al. Disturbed ratio of erythrocyte and plasma S-adenosylmethionine/S-adenosylhomocysteine in peripheral arterial occlusive disease. Atherosclerosis 154, 147–154 (2001).
Gu, X. et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818 (2017).
Zhang, H. & Ge, Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ. Cardiovasc. Genet. 4, 711 (2011).
McCorvie, T. J. et al. Inter-domain communication of human cystathionine beta-synthase: structural basis of S-adenosyl-L-methionine activation. J. Biol. Chem. 289, 36018–36030 (2014).
Patel, D., Kopec, J., Fitzpatrick, F., McCorvie, T. J. & Yue, W. W. Structural basis for ligand-dependent dimerization of phenylalanine hydroxylase regulatory domain. Sci. Rep. 6, 23748 (2016).
Jaffe, E. K. Impact of quaternary structure dynamics on allosteric drug discovery. Curr. Top. Med. Chem. 13, 55–63 (2013).
Sun, D. F. et al. Knock-down of methylenetetrahydrofolate reductase reduces gastric cancer cell survival: an in vitro study. Cell. Biol. Int. 32, 879–887 (2008).
Stankova, J., Lawrance, A. K. & Rozen, R. Methylenetetrahydrofolate reductase (MTHFR): a novel target for cancer therapy. Curr. Pharm. Des. 14, 1143–1150 (2008).
Winter, G., Lobley, C. M. & Prince, S. M. Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 69, 1260–1273 (2013).
Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 (2007).
Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 (2006).
McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 (2007).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Rummel, T. et al. Intermediate hyperhomocysteinaemia and compound heterozygosity for the common variant c.677CT and a MTHFR gene mutation. J. Inherit. Metab. Dis. 30, 401 (2007).
Shapiro, S. K. & Ehninger, D. J. Methods for the analysis and preparation of adenosylmethionine and adenosylhomocysteine. Anal. Biochem. 15, 323–333 (1966).
Niesen, F. H., Berglund, H. & Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 (2007).
Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 25, 495–503 (1992).
Chalk R. in Heterologous Gene Expression in E. coli: Methods and Protocols (ed. Burgess-Brown N. A.) 373–395 (Humana Press, New York City, 2017).
Testa, L., Brocca, S. & Grandori, R. Charge-surface correlation in electrospray ionization of folded and unfolded proteins. Anal. Chem. 83, 6459–6463 (2011).
Svergun, D., Barberato, C. & Koch, M. H. J. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 (1995).
Acknowledgements
This work was supported by the Olga-Mayenfisch Stiftung (to D.S.F.), the Rare Disease Initiative Zurich, a Clinical Research Priority Program from the University of Zurich (to D. S. F. and M. R. B.) and the Swiss National Science Foundation (SNSF 31003A_156907) (to M.R.B.). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, MSD, Merck KGaA, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda and Wellcome Trust [106169/ZZ14/Z]. We thank the Diamond Light Source for beamtime, and the staff of beamline I04-1 for assistance with data collection. We thank Merima Forny for assistance with sequencing.
Author information
Authors and Affiliations
Contributions
D.S.F., M.R.B. and W.W.Y. conceived of the study. D.S.F., J.K. and E.R. cloned, purified and crystallized MTHFR constructs. They also performed SAM binding and dephosphorylation assays. J.K., A.O. and G.A.B. performed X-ray structure determination. G.A.B. performed and analysed the SAXS measurement. T.S. and S.L. performed and analysed the activity assays. R.C. and O.B. performed and analysed the mass spectrometry assays. D.S.F. and W.W.Y. wrote the manuscript with editing and proofreading from the other co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Froese, D.S., Kopec, J., Rembeza, E. et al. Structural basis for the regulation of human 5,10-methylenetetrahydrofolate reductase by phosphorylation and S-adenosylmethionine inhibition. Nat Commun 9, 2261 (2018). https://doi.org/10.1038/s41467-018-04735-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-04735-2
- Springer Nature Limited