
Abstract
Damage to the ribosome or an imbalance in protein biosynthesis can lead to some human diseases, such as diabetic retinopathy (DR) and other eye diseases. Here, we reported that the kri1l gene was responsible for retinal development. The kri1l gene encodes an essential component of the rRNA small subunit processome. The retinal structure was disrupted in kri1l mutants, which resulted in small eyes. The boundaries of each layer of cells in the retina were blurred, and each layer of cells was narrowed and decreased. The photoreceptor cells and Müller glia cells almost disappeared in kri1l mutants. The lack of photoreceptor cells caused a fear of light response. The development of the retina started without abnormalities, and the abnormalities began two days after fertilization. In the kri1l mutant, retinal cell differentiation was defective, resulting in the disappearance of cone cells and Müller cells. The proliferation of retinal cells was increased, while apoptosis was also enhanced in kri1l mutants. γ-H2AX upregulation indicated the accumulation of DNA damage, which resulted in cell cycle arrest and apoptosis. The kri1l mutation reduced the expression of some opsin genes and key retinal genes, which are also essential for retinal development.
Similar content being viewed by others


Introduction
In the vertebrate central nervous system (CNS), neural retina neurogenesis is a very good system because the retinal structure and developmental mechanisms are highly conserved [1]. Zebrafish (Danio rerio) is a type of spinal model organism. At 72 hpf (hours post fertilization, hpf), the retina of zebrafish is close to maturity, and the retinal structure and function are similar to those of the human retina. The mature zebrafish retina is composed of three separate nuclear layers by two reticular layers. From the outside to the inside, they are the outer nuclear layer, the outer reticular layer, the inner nuclear layer, the inner reticular layer and the ganglion cell layer, and photoreceptor cell bodies exist in the outer nuclear layer. Amacrine glial cells, horizontal glial cells, and Müller glial cells occupy the inner nuclear layer. Ganglion cells exist in the ganglion cell layer, and synapsis between these nuclear layers occurs at the plexiform layers [2].
The ribosome is a highly conserved ribonucleoprotein complex that is the main center of mRNA and protein quality control [3]. Ribosomes are important and highly complex machines responsible for the synthesis of proteins in all cell growth processes, and diseases caused by ribosome biosynthesis disorders are called ribosomal diseases, such as diabetic retinopathy (DR), glaucoma, cataracts and other eye diseases [4,5,6,7]. In addition, ribosome biogenesis must respond rapidly to environmental cues mediated by internal and cell surface receptors or stress (oxidative stress, DNA damage, amino acid depletion, etc.) [8]. Impaired ribosomal biosynthesis can cause DNA damage [9]. DNA damage may be caused by various endogenous or exogenous stresses, including oxidative stress, telomere erosion, carcinogenic mutation, genotoxic stress, and metabolic stress.
This study reported that kri1l was essential for retinal development. Kri1l is an important part of the ribosomal 45 S rRNA cleavage complex and is responsible for the cleavage of ribosomal 18 S rRNA [10]. Kri1l deletion causes disordered ribosome biosynthesis through the accumulation of DNA damage, resulting in cell cycle arrest and apoptosis. At the same time, the kri1l mutation reduced the expression of certain opsin genes and key retinal genes.
Results
Mutant kri1l causes retinal morphological defects
The kri1l−/− mutant (kri1lcas002) was obtained from a large-scale forward genetics screen with ENU-mutagenized in zebrafish. And we carried out positional cloning and found a mutantion in kri1l (also named kri1) gene responsible for the phenotype [11]. In eukaryotic cells, a ribonucleoprotein (RNP) called small subunit (SSU) processome takes charge of generating mature 18 S rRNA and assembling small ribosomal subunit. Kri1l is a component of the SSU complex, loss of Kri1l results in instability of 18 S rRNA precursor and dramatic reducation of mature 18 S rRNA. Kri1l is essential for the formation of polysome and 40 S ribosome subunits. The retina of zebrafish is close to maturity at 72 hpf, so 72 hpf after fertilization is the best time to observe retinal development. Under an optical microscope, we found that the kri1l mutation caused the mutant eyes to decrease compared to wild-type embryos (Fig. 1A–D). After measuring the eye area of kri1l mutants at 3 dpf, 4 dpf, and 5 dpf, it was found that the eye area of the kri1l mutant was smaller than that of the wild-type embryo (Fig. 1G). To further explore changes in the retina, cryosections of the eyes were subjected to hematoxylin-eosin staining (HE). The results showed that the boundaries of each retinal cell layer became fuzzy, and each layer cell became narrower in the kri1l mutant, indicating that the cell number was reduced in each retinal layer (Fig. 1E, F). We counted the area of each layer cells in the retina, and the total area in each layer significantly decreased (Fig. 1H–L). Hoechst staining also indicated a significant decrease in the number of cells in each layer. These data indicated the retinal development was severely disrupted in kri1l−/−.

A–D Light microscope image of zebrafish embryos at 4 dpf. Scale bar, 100 μm. G Statistical analysis of the eye surface area in sibling and kri1l−/− mutants. Error bars represent the standard deviation (SD). ***, P < 0.001. The black arrows indicate the eye. E, F H&E staining of frozen embryo eye sections at 4 dpf. Scale bar, 50 μm. The zoom section had shown in E′–F′. The yellow arrow indicated the ONL (outer nuclear layer). The red arrow indicated the OPL (Outer plaxiform layer). The blue arrow indicated the INL (inner nuclear layer). The orange arrow indicated the IPL (Inner plaxiform layer). The green arrow indicated the GCL (ganglion cell layer). H–L Statistical analysis of the indicated area in sibling and kri1l−/− mutants.
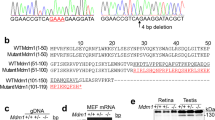
After immunofluorescence staining and photographing, all embryos were extracted for genomic DNA and genotyped by sequencing. There was a 38 bp deletion in the cDNA of kri1l gene in mutants (Fig. S1A). Genomic DNA sequencing of kri1l gene revealed that a consensus splicing donor site at the boundary between exon1 and intron1 was disrupted by a T-to-G transitionin in mutants (Fig. S1B). This transition T-to-G spliced from the earlier GT position in exon1 and yielded an alternative splicing transcript with a frame shift (Fig. S1C). We designed primers (which located on exon 1 and exon 2), and amplified from genomic DNA, and found that the product in the mutants was 192 bp, significantly smaller than that in wild-type 230 bp (Fig. S1D).
kri1l is required for retinal differentiation
To determine the differentiation patterns in kri1l−/− mutants, we examined differentiated retinal neurons (Zpr-1 for photoreceptor cells [12]) and Müller glial cells (GS, markers for Müller glia cells [13]). We found these differentiated retinal neurons were substantially reduced, including Zpr-1 (Fig. 2A–F) and GS (Fig. 2H–M). The number of Zpr-1+ and GS+ cells was significantly reduced in kri1l−/− mutants (Fig. 2G, N).

A–F Representative Zpr-1 (for photoreceptor cells) immunofluorescence images for frozen embryo eye sections from sibling A–C and kri1l−/− mutants D–F at 4 dpf. G Statistical analysis of Zpr-1+ cells in sibling and kri1l−/− mutants. H–M Representative GS (for Müller glial cells) immunofluorescence images for frozen embryo eye sections from sibling H–J and kri1l−/− mutants K–M at 4 dpf. N Statistical analysis of Müller glial cells in sibling and kri1l−/− mutants. Error bars represent the standard deviation (SD). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bar, 50 μm.
Another retinal differentiation marker Islet1 (Islet1 for inner nuclear layer cells), were decreased (Fig. 3A–G). The inner nuclear layer contains the cell body of bipolar cells, horizontal cells, and amacrine cell cells. And the migration of retinal neurons occur from the inner side to the outer side, starting from inner nuclear layer cells. The Islet1 positive cells decreased suggested nearly all differentiated retinal neurons were deficiency.

A–F Representative Islet1 (for inner nuclear layer cells) immunofluorescence images for frozen embryo eye sections from sibling A–C and kri1l−/− mutants D–F at 4 dpf. G Statistical analysis of islets + cells in sibling and kri1l−/− mutants. Error bars represent the standard deviation (SD). ***, P < 0.001. Scale bar, 50 μm.
Retinal lamination is initiated by the migration of postmitotic neurons to the appropriate cell layer. When neurons migrate to the appropriate retinal region, they mature and establish synapses between different cell layers and differentiate into seven types of retinal cells: Müller glia cells, bipolar cells, cone photoreceptors and so on. Multipotent RPCs (retinal progenitor cells) can differentiate into all types of retinal neurons [2, 14]. To understand when the differentiation of retinal cells initiated failure, we traced retinal progenitor cell (sox2, markers for RPCs) expression at sequential developmental time points. Sox2 expression was normal in the retina at 36 hpf (Fig. 4A–F). The initial decrease in sox2 expression was detectable in the retinal region at 2 dpf (Fig. 4G–L). To further confirm these results, Tg(HuC:eGFP) living embryos were observed at 2 dpf, which labeled neurons. Consistently, HuC:eGFP+ cells were dramatically reduced in kri1l−/− retinas (Fig. 4M–R). Above all, the retinal differentiation defects initiated from 2dpf, and RPCs could not differentiate into various retinal lineages during neurogenesis in the absence of kri1l.

A–F The distributions of Sox2+ cells (markers for RPC) in whole-mount retinas from WT and kri1l−/− mutants at 36 hpf. G–L The distributions of Sox2+ cells (markers for RPC) in whole-mount retinas from WT and kri1l−/− mutants at 2 dpf. M–R The distributions of HuC:EGFP (postmitotic neurons)-labeled cells in whole-mount retinas from WT and kri1l−/− transgenic zebrafish at 2 dpf. The dashed circles show the eyes and lenses. Scale bar, 100 μm.
Defects in photoreceptor cells in the retina caused a fear of light response in kri1l mutants
To further explore the response of the kri1l mutant to light stimulus in the absence of photoreceptor cells, we performed alternating light and dark light cycles to stimulate equal behavioral experiments. The total distance and swimming speed of kri1l−/− were all lower than those of wild-type embryos (Fig. 5A). The total distance of the kri1l mutant in the dark was less than that in the light (Fig. 5A). The kri1l mutant showed an increase in average speed and startle within 5 min of light. After turning off the light source, the speed quickly decreased, but the average speed of wild-type embryos remained basically unchanged under different light conditions (Fig. 5B). These data suggested that the swimming speed of mutant larvae exhibited an uptick in the light period, indicating that the mutant larvae showed a fear of light response.

A The total distance of the larvae during the 5 min dark and 5 min light period. B The average swimming speed of the larvae during the 5 min dark and 5 min light period. * are compared between WT and kri1l−/− mutants, **, P < 0.01; ***, P < 0.001; # are compared between dark period and light period in kri1l−/− mutants, #, P < 0.05; ##, P < 0.01; Results are presented as the mean ± SD.
Proliferation of retinal cells increased and apoptosis was also enhanced in kri1l mutants
To observe the mechanism of the decrease in retinal cells, we performed cell proliferation and apoptosis in retinas by pH3 and EdU immunofluorescence and TUNEL assays. pH3 (phosphorylation at Ser 10 of histone H3) immunofluorescence and EdU staining were used to detect cell proliferation. pH3 staining was enhanced, which can mark cell proliferation in the G2/M division stage [15] (Fig. 6A–G). EdU labeling also increased, which was used to detect cell proliferation in the S stage [16] (Fig. 6H–N). The pH3 and EdU staining all indicated the cell proliferation was elevated in mutants.

A–F pH3 (marked for G2/M-phase cells) staining in frozen embryo eye sections of sibling A–C and kri1l−/− mutants D–F at 4 dpf. G The pH3+ cell quantitative results are summarized in A–F. H–M EdU (marked for S-phase cells) staining in frozen embryo eye sections of sibling H–J and kri1l−/− mutants K–M at 4 dpf. N The EdU+ cell quantitative results are summarized in H–M. Error bars represent the standard deviation (SD). ***P ≤ 0.001 (Student’s t test). Scale bars, 50 μm.
TUNEL staining detects cell apoptosis [17, 18]. The results showed that the TUNEL-positive signal was noticeably elevated in the kri1l mutant, suggesting the activation of apoptosis (Fig. 7A–G). To define the apoptosis, we looked at additional markers of apoptosis such as cleaved caspase 3 and cleaved caspase 9. The number of cleaved caspase 3 + cells (Fig. 8A–G) and cleaved caspase 9 + cells (Fig. 8H–N) in kri1l−/− mutants were significantly increased. Together with TUNEL results (Fig. 7), all these results suggested that the apoptosis level was elevated in kri1l−/− mutants.

A–F TUNEL staining in frozen embryo eye sections of sibling A–C and kri1l−/− mutants D–F at 4 dpf. G Quantitative results are summarized. Error bars represent the standard deviation (SD). ***, P ≤ 0.001 (Student’s t test). Scale bars, 50 μm.

A–F Representative cleaved caspase3 immunofluorescence images for frozen embryo eye sections from sibling A–C and kri1l−/− mutants D–F at 4 dpf. The zoom section had shown in A′-F′. G Statistical analysis of cleaved caspase3 + cells in sibling and kri1l−/− mutants. H–M Representative cleaved caspase9 immunofluorescence images for frozen embryo eye sections from sibling H–J and kri1l−/− mutants K–M at 4 dpf. The zoom section had shown in H′–M′. N Statistical analysis of cleaved caspase9 + in sibling and kri1l−/− mutants. Error bars represent the standard deviation (SD). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bar, 50 μm.
Accumulation of DNA damage resulted in cell cycle arrest and apoptosis
Many studies have shown that DNA damage affects cell proliferation and apoptosis, hindering cell cycle progression [19,20,21]. We suspected that there was DNA damage in the kri1l mutant. To test this hypothesis, we performed DNA damage detection in the kri1l mutant. γ-H2AX is phosphorylated H2AX, and the phosphorylation site is located at serine 139. The expression of the γ-H2AX gene can detect DNA break sites [22, 23]. The results showed that the γ-H2AX signal (Fig. 9A–G) was enhanced in the retinas of the kri1l mutant.

A–F Immunofluorescence analysis using the anti-γ-H2AX antibody in sibling A–C and kri1l−/− mutant D–F retinas at 4 dpf. The zoom section had shown in A′–F′. G Statistical analysis of γ-H2AX + cells in sibling and kri1l−/− mutants. Scale bar, 50 μm.
The expression of some opsin genes and key retinal genes was downregulated in kri1l mutants
The mRNA levels of five opsin genes and some key retinal genes were also examined by qRT‒PCR tests in the kri1l mutant. The mRNA levels of the zfrho, zfred, zfgr1, zfuv and zfblue genes, which encode rhodopsin, red, green, ultraviolet and blue opsins, respectively, were significantly and consistently downregulated. The gdf6a, rx1, rx2, rx3, pax6a and pax6b genes are related to microphthalmia. The rp2, cerkl, myo7aa, ush1c and pcdh15a genes are related to retinitis pigmentosa. The cdipt gene is related to cataracts. The slc45a2 and lrmda genes are associated with albinism [2]. The gucy2f gene is related to Leber’s congenital amaurosis (LCA). The vhl gene is related to the development of retinal blood vessels. The results showed that the mRNA levels of the opsin genes were reduced in the kri1l mutant (Fig. 10A). The mRNA expression levels of genes related to microphthalmia (Fig. 10B), retinitis pigmentosa (Fig. 10C), cataract (Fig. 10D), ocular albinism (Fig. 10D), Leber’s congenital amaurosis (Fig. 10D), and retinal vascular development (Fig. 10D) were all downregulated in the kri1l mutant. Therefore, the downregulation of expression of some visual protein genes and key retinal genes in the kri1l mutant may lead to retinal dysplasia.

The relative expression of in sibling and kri1l−/− mutant embryos at 4 dpf. A Relative expression of opsin gene. B Relative expression of microphthalmia diseases gene. C Relative of RPE disease gene mRNA level. D Relative of other eye diseases gene mRNA level. Error bars represent standard deviation (SD). **P ≤ 0.01; ***P ≤ 0.001.
Discussion
This study showed that the kri1l mutation caused abnormal retinal development, with fuzzy boundary retinal cell layers, and each layer cell became narrower and smaller in the kri1l mutant. Retinal differentiation was damaged because photoreceptor cells and Müller glial cells almost disappeared in kri1l mutants. The lack of photoreceptor cells caused a fear of light response. It might be the accumulation of DNA damage that resulted in cell cycle arrest and apoptosis. At the same time, the kri1l mutation reduced the expression of certain opsin genes and key retinal genes. The working model for Kri1l dysfunction abnormal retinal development was in Fig. S2.
Kri1l gene research first appeared in 2000 and is involved in the splicing process of 18 S rRNA, and yeast mutant strains that lack the kri1 gene exhibit slow growth [10, 24]. Subsequent reports mentioned that loss of kri1l caused ribosomal biogenesis defects, accumulation of misfolded proteins and activation of PERK-eif2a signaling. These deficiencies subsequently hyperactivate autophagy and ultimately lead to the inhibition of HSPC proliferation [11]. In addition, all embryos homozygous for the kri1l mutation died within 5–10 days of fertilization. The evidence could attest to the importance of kri1l development. In human diseases, the KRI1 mutation may be relevant with severe iron deficiency anemia [25]. And KRI1 were significantly correlated with esophageal carcinoma tumor location, lymph node metastasis, and age of patients. KRI1 had the highest mutation frequency from the TCGA database [26].
Our results suggested that when kri1l was mutated, the structure of the retina was destroyed, leading to the appearance of ommatidia and fuzzy boundary retinal cell layers, and each layer cell became narrower and smaller. Studies have reported that plk1, vps28, psmd2, ran, sec13 [27], ccdc94 [28], and tln1 [29] mutations cause ommatidia in zebrafish embryos. The plk1 mutant had abnormal retinal development and did not form a normal retinal morphology. The vps28 mutant had slightly smaller eyes, but the retinal stratification was normal; the psmd2 and ran mutants had damaged retinal stratification [30]. These retinal defects were very similar to our results (Fig. 1).
Photoreceptor cells (cones and rods) are used to receive light signals, process the signals and transmit them to other retinal nerve cells. When the retina is lost, Müller cells re-enter the cell cycle to proliferate and differentiate into damaged nerve cells, thereby repairing the damage to the retina [31,32,33]. Previous studies have reported cone differentiation defects in her9 and lca535 [Histologic analysis Briefly, the collected cryosections were fixed in 4% paraformaldehyde for 10 min and washed in water for 2 min. The cryosections were stained with filtered hematoxylin solution (Beyotime, C0105) for 8 min at room temperature (RT). Following a wash in distilled water for 2 min, the sections were treated with 0.3% hydrochloric acid ethanol solution for 2 s, washed with water for 2 min, counterstained in eosin dye for approximately 30 s at RT, placed in 70%, 95%, and 100% ethanol dehydrate in the solution for 10 s, and mounted on slides with antifade mounting medium. Finally, high-resolution images of the H&E-stained sections were obtained under a microscope (Nikon, SMZ18). N ≥ 6, the experiment was repeated at least three times. Different groups of zebrafish embryos were placed into a 24-well plate, and the zebrafish embryo movement behavior instrument was controlled to collect 30 min of motion video. Among them, there was no light for the first 5 min, and there was light for 5 min. The light and dark cycles were repeated 3 times. EthoVision XT software was used to export motion speed parameters. N ≥ 24, the experiment was repeated at least three times. The cryosections were stained using an in situ cell death detection kit. The cryosections were circled with a hydrophobic pen, fixed in 4% paraformaldehyde for 20 min, treated with acetone at -20 °C for 7 min and 1% sodium citrate in PBS for 1 h, stained with a TUNEL staining kit, and stained with 0.1% Tween’s PBS to terminate the reaction. Anti-quenching reagent was used to seal the coverslip. N ≥ 6, the experiment was repeated at least three times. The cryosections were treated with acetone at -20 °C for 7 min. Samples were blocked with blocking buffer (10% goat serum in PBS with 0.5% Triton X-100) for 2 h at room temperature and incubated with primary antibody overnight. Primary antibodies included Zpr1 (abcam, ab174435, 1:300), GS (Merck, MAB302, 1:300), phosphorylated histone H3 (CST, 3377, 1:500), γ-H2AX (abcam, ab11174, 1:300), Istel1 (GeneTex, GTX102807, 1:200), caspase 3 (CST, #9661, 1:300), and caspase 9 (abcam, ab202068, 1:500). Secondary antibodies conjugated with Alexa Fluor488 were used at a 1:500 dilution. The reaction was terminated with 0.1% Tween in PBS. Hoechst (Beyotime, C1022) dye was used to stain the nucleus (1:2500), and the slides were mounted with antifade mounting medium. Images were obtained with a confocal microscope (Nikon, A1+ Confocal Microscope). N ≥ 6, the experiment was repeated at least three times. The embryos were treated with 2 mM 5-ethynyl-2-deoxyuridine (EdU) for 20 min at 4 °C and then washed 3 times with egg water. The embryos were transferred to egg water in an incubator at 28.5 °C. After 4 h, the embryos were fixed in 4% paraformaldehyde overnight, dehydrated in a methanol gradient overnight and rehydrated. Zebrafish embryos were used for frozen sectioning, and then the Click-iT Plus EdU Imaging Kit (Invitrogen, C10640) was used to process the sections for 30 min. The reaction was terminated using PBS with 0.1% Tween. N ≥ 6, the experiment was repeated at least three times. The Tg(HuC:eGFP) live embryos were anesthetized with 0.08% tricaine and mounted in 0.1% low melting point agarose for imaging with a Nikon A1+ confocal microscope (under a 20× water-immersion objective). N ≥ 24, the experiment was repeated at least three times. To detect sox2 mRNA, embryos were first hybridized with the DIG-labeled antisense sox2 RNA probe, incubated at 4 °C overnight with a peroxidase (POD)-conjugated anti-DIG antibody (1:500; Roche), and stained with Alexa Fluor cy3-conjugated tyramide as substrate (PerkinElmer). Images were obtained with a confocal microscope (Nikon, A1+ Confocal Microscope). The experiment was repeated at least three times. N ≥ 24, the experiment was repeated at least three times. Total RNA was extracted from 16 zebrafish embryos using Trizol reagent. Reverse transcription was performed with the Thermo Scientific RNA Reverse Transcription Kit. 2× PCR Mix (TaKaRa, Premix Ex Taq) containing SYBR Green I was used for the real-time quantitative PCR analysis with the Roche Applied Science Fast Real-Time PCR System. The corresponding gene primers are shown in Table S1. The experiment was repeated at least three times. GraphPad Prism 8 software was used to perform t tests for all statistical analyses. P < 0.05 indicated a statistically significant difference, and all the values are the mean ± standard deviation. Each experiment was repeated at least three times.Locomotion analysis in larval zebrafish
Terminal deoxynucleotidyl transferase dUTP Nick End Labeling (TUNEL) staining
Immunofluorescence
Detection of cell proliferation by EdU staining
Live imaging of Tg(HuC:eGFP) embryos
Whole mount in situ hybridization
Quantitative real-time polymerase chain reaction (qRT‒PCR)
Statistical analyses
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Centanin L, Wittbrodt J. Retinal neurogenesis. Development. 2014;141:241–4.
Richardson R, Tracey-White D, Webster A, Moosajee M. The zebrafish eye-a paradigm for investigating human ocular genetics. Eye (Lond). 2017;31:68–86.
Brandman O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol. 2016;23:7–15.
Farley KI, Baserga SJ. Probing the mechanisms underlying human diseases in making ribosomes. Biochem Soc Trans. 2016;44:1035–44.
Zhang SX, Ma JH, Bhatta M, Fliesler SJ, Wang JJ. The unfolded protein response in retinal vascular diseases: implications and therapeutic potential beyond protein folding. Prog Retin Eye Res. 2015;45:111–31.
Kroeger H, Chiang WC, Felden J, Nguyen A, Lin JH. ER stress and unfolded protein response in ocular health and disease. Febs j. 2019;286:399–412.
Zhang K, Zhu X, Lu Y. The proteome of cataract markers: focus on crystallins. Adv Clin Chem. 2018;86:179–210.
Piazzi M, Bavelloni A, Gallo A, Faenza I. Signal transduction in ribosome biogenesis: a recipe to avoid disaster. Int J Mol Sci. 2019;20:2718.
Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, et al. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature. 2018;554:112–7.
Sasaki T, Toh EA, Kikuchi Y. Yeast Krr1p physically and functionally interacts with a novel essential Kri1p, and both proteins are required for 40S ribosome biogenesis in the nucleolus. Mol Cell Biol. 2000;20:7971–9.
Jia XE, Ma K, Xu T, Gao L, Wu S, Fu C, et al. Mutation of kri1l causes definitive hematopoiesis failure via PERK-dependent excessive autophagy induction. Cell Res. 2015;25:946–62.
Larison KD, Bremiller R. Early onset of phenotype and cell patterning in the embryonic zebrafish retina. Development. 1990;109:567–76.
Lust K, Wittbrodt J. Activating the regenerative potential of Müller glia cells in a regeneration-deficient retina. Elife. 2018;7:e32319.
Alves CH, Pellissier LP, Wijnholds J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog Retin Eye Res. 2014;40:35–52.
Lin Q, Mei J, Li Z, Zhang X, Zhou L, Gui JF. Distinct and cooperative roles of amh and dmrt1 in self-renewal and differentiation of male germ cells in Zebrafish. Genetics. 2017;207:1007–22.
Flomerfelt FA, Gress RE. Analysis of cell proliferation and homeostasis using EdU labeling. Methods Mol Biol. 2016;1323:211–20.
Gorczyca W, Traganos F, Jesionowska H, Darzynkiewicz Z. Presence of DNA strand breaks and increased sensitivity of DNA in situ to denaturation in abnormal human sperm cells: analogy to apoptosis of somatic cells. Exp Cell Res. 1993;207:202–5.
Lozano GM, Bejarano I, Espino J, González D, Ortiz A, García JF, et al. Relationship between caspase activity and apoptotic markers in human sperm in response to hydrogen peroxide and progesterone. J Reprod Dev. 2009;55:615–21.
Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. 2014;20:967–77.
da Luz Dias R, Basso B, Donadio MVF, Pujol FV, Bartrons R, Haute GV, et al. Leucine reduces the proliferation of MC3T3-E1 cells through DNA damage and cell senescence. Toxicol In Vitro. 2018;48:1–10.
Luo T, Yu Q, Zou H, Zhao H, Gu J, Yuan Y, et al. Role of poly (ADP-ribose) polymerase-1 in cadmium-induced cellular DNA damage and cell cycle arrest in rat renal tubular epithelial cell line NRK-52E. Environ Pollut. 2020;261:114149.
Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68.
Zheng S, Lan P, Liu X, Ye K. Interaction between ribosome assembly factors Krr1 and Faf1 is essential for formation of small ribosomal subunit in yeast. J Biol Chem. 2014;289:22692–703.
Huettmann C, Stelljes M, Sivalingam S, Fobker M, Vrachimis A, Exler A, et al. Iron deficiency caused by intestinal iron loss-novel candidate genes for severe anemia. Genes (Basel). 2021;12:1869.
Wang J, Guo Z, Sun F, Xu T, Wang J, Yu J. A novel ferroptosis-related gene signature to predict prognosis of esophageal carcinoma. J Oncol. 2022;2022:7485435.
Niu X, Hong J, Zheng X, Melville DB, Knapik EW, Meng A, et al. The nuclear pore complex function of Sec13 protein is required for cell survival during retinal development. J Biol Chem. 2014;289:11971–85.
Sorrells S, Carbonneau S, Harrington E, Chen AT, Hast B, Milash B, et al. Ccdc94 protects cells from ionizing radiation by inhibiting the expression of p53. PLoS Genet. 2012;8:e1002922.
Wu Q, Zhang J, Koh W, Yu Q, Zhu X, Amsterdam A, et al. Talin1 is required for cardiac Z-disk stabilization and endothelial integrity in zebrafish. Faseb j. 2015;29:4989–5005.
Hu R, Huang W, Liu J, ** M, Wu Y, Li J, et al. Mutagenesis of putative ciliary genes with the CRISPR/Cas9 system in zebrafish identifies genes required for retinal development. Faseb J. 2019;33:5248–56.
Salesse C. Physiology of the visual retinal signal: From phototransduction to the visual cycle. J Fr Ophtalmol. 2017;40:239–50.
Hamon A, Roger JE, Yang XJ, Perron M. Müller glial cell-dependent regeneration of the neural retina: An overview across vertebrate model systems. Dev Dyn. 2016;245:727–38.
Dyer MA. Regulation of proliferation, cell fate specification and differentiation by the homeodomain proteins Prox1, Six3, and Chx10 in the develo** retina. Cell Cycle. 2003;2:350–7.
Qu Z, Yimer TA, **e S, Wong F, Yu S, Liu X, et al. Knocking out lca5 in zebrafish causes cone-rod dystrophy due to impaired outer segment protein trafficking. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2694–705.
Coomer CE, Wilson SG, Titialii-Torres KF, Bills JD, Krueger LA, Petersen RA, et al. Her9/Hes4 is required for retinal photoreceptor development, maintenance, and survival. Sci Rep. 2020;10:11316.
Aboshiha J, Dubis AM, Carroll J. The cone dysfunction syndromes. Br J Ophthalmol. 2016;100:115–21.
Fischer MD, Michalakis S, Wilhelm B, Zobor D, Muehlfriedel R, Kohl S, et al. Safety and vision outcomes of subretinal gene therapy targeting cone photoreceptors in achromatopsia: a nonrandomized controlled trial. JAMA Ophthalmol. 2020;138:643–51.
Li J, Liu F, Lv Y, Sun K, Zhao Y, Reilly J, et al. Prpf31 is essential for the survival and differentiation of retinal progenitor cells by modulating alternative splicing. Nucleic Acids Res. 2021;49:2027–43.
Novales Flamarique I, Sayed Ahmed A, Cheng CL, Molday RS, Devlin RH. Growth hormone regulates opsin expression in the retina of a salmonid fish. J Neuroendocrinol. 2019;31:e12804.
Funding
This work was supported by the National Natural Science Foundation of China (81901918, 81660204), Inner Mongolia Science Foundation (2019MS08060, 2021LHMS08022), Inner Mongolia High School Science Research Foundation (NJZY23093, NJZZ23106), Local science and technology projects guided by the central government (2020ZY0040), CAS “Light of West China” Program to XEJ, Inner Mongolia Talent Development Fund to XEJ (2022), Innovation Team of Baotou Medical College (bycxtd-04), Innovative and Entrepreneurial Talents in the “Prairie Talents” Project of Inner Mongolia (Q2017047), Baotou Medical College Foundation (BSJJ201804), and college student project (HL2021005).
Author information
Authors and Affiliations
Contributions
XEJ, CJZ, WJP, and WZ designed research, analyzed data, and wrote the manuscript; RZ, YBX, JJS, MTT, and YC performed experiments; SYJ, XLL assisted with the experiments; WX, RGBD, and GS gave suggestions for experiment design.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Zhang, R., Sun, J., **e, Y. et al. Mutant kri1l causes abnormal retinal development via cell cycle arrest and apoptosis induction. Cell Death Discov. 10, 251 (2024). https://doi.org/10.1038/s41420-024-02022-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-024-02022-2
- Springer Nature Limited