
Abstract
Metabolic reprogramming is a hallmark of human malignancies. Dysregulation of glutamine metabolism is essential for tumorigenesis, microenvironment remodeling, and therapeutic resistance. Based on the untargeted metabolomics sequencing, we identified that the glutamine metabolic pathway was up-regulated in the serum of patients with primary DLBCL. High levels of glutamine were associated with inferior clinical outcomes, indicative of the prognostic value of glutamine in DLBCL. In contrast, the derivate of glutamine alpha-ketoglutarate (α-KG) was negatively correlated with the invasiveness features of DLBCL patients. Further, we found that treatment with the cell-permeable derivative of α-KG, known as DM-αKG, significantly suppressed tumor growth by inducing apoptosis and non-apoptotic cell death. Accumulation of a-KG promoted oxidative stress in double-hit lymphoma (DHL), which depended on malate dehydrogenase 1 (MDH1)-mediated 2-hydroxyglutarate (2-HG) conversion. High levels of reactive oxygen species (ROS) contributed to ferroptosis induction by promoting lipid peroxidation and TP53 activation. In particular, TP53 overexpression derived from oxidative DNA damage, further leading to the activation of ferroptosis-related pathways. Our study demonstrated the importance of glutamine metabolism in DLBCL progression and highlighted the potential application of α-KG as a novel therapeutic strategy for DHL patients.

Similar content being viewed by others
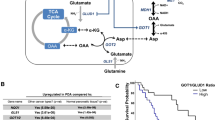

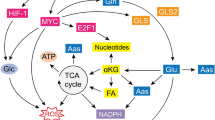
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin’s lymphoma (NHL), featured by heterogeneity, aggressiveness, and poor prognosis [1, 2]. With the emergence of anti-CD20 monoclonal antibodies, the survival rates for DLBCL patients have improved significantly. However, approximately 40% of cases progress to relapsed/refractory DLBCL [3]. Recently, molecular heterogeneity of DLBCL has been identified as a major factor interfering with clinical treatment responses [4], especially in double-hit lymphoma (DHL) and triple-hit lymphoma (THL) [5, 6]. Investigation of new therapeutic strategies is an urgent need to guide clinical practice.
As a prominent hallmark of cancer, metabolic reprogramming represents the phenotypic outcome and environmental influences on the molecular function of tumor cells [7]. Reorganization of tumor metabolism is characterized by increased assumption of various nutrients, including glutamine [8]. Glutamine is a vital metabolic fuel that is essential for ATP production and precursors biosynthesis [9]. Intracellular glutamine is converted into glutamate and alpha-ketoglutarate (α-KG) to support the tricarboxylic acid (TCA) cycle [10]. Glutamine metabolism also promotes nucleotides and protein biosynthesis to meet the demand for rapid tumor growth [11, 12]. In addition to energy supply, activation of glutamine metabolism contributes to counteracting the adverse effects of ammonia on tumor cells [13]. Therefore, glutamine metabolism plays an important role in tumor growth, microenvironment regulation, and drug resistance [14]. A recent study demonstrated that metabolic reprogramming-mediated glutaminolysis was associated with Bruton’s tyrosine kinase inhibitor (BTKi) resistance in mantle cell lymphoma (MCL) [15], suggesting the promise of targeting metabolism in hematological tumors.
As the major derivative of glutamine, a steady level of α-KG is essential for various biological processes [16, 17], while accumulation of α-KG conversely has tumor-suppressing effects. Elevated level of α-KG suppresses tumorigenesis by switching glucose metabolism [18, 19] and hinders tumor progression by driving tumor cell differentiation [20]. In addition, accumulated α-KG blocks tumor growth by inducing gasdermin C (GSDMC)-mediated pyroptosis [17], indicating that cell death induction by α-KG may be considered a potential strategy for cancer. Consistently, α-KG plays an important role in DLBCL, which depends on the functions of α-KG in promoting TCA process [21]. Since α-KG-induced cell death has yet to be described in DLBCL, investigating the mechanisms of α-KG will hope to provide novel therapeutic strategies for DLBCL patients.
Non-apoptotic cell death is evolving into a new cancer treatment strategy due to its influence on tumor progression and treatment response [22, 23]. As a novel form of non-apoptotic cell death, ferroptosis is derived from iron overload and lipid reactive oxygen species (ROS) accumulation [24]. In contrast to cell apoptosis, ferroptosis has specific morphological features, including shrinkage of mitochondria, reduction of mitochondrial cristae, and condensation of the internal membrane [25, Intracellular α-KG was converted to 2-HG by MDH1, further promoting ROS production. ROS accumulation promoted lipid membrane peroxidation and initiated oxidative DNA damage. TP53 overexpression derived from oxidative DNA damage participated in regulating the ferroptosis pathways. TP53-mediated ferroptosis pathways, together with lipid peroxidation, further led to the generation of ferroptosis.
Metabolic reprogramming is essential for maintaining the biological functions of tumor cells and immune cells [7, 50]. As a kind of aggressive B-cell lymphoma, DLBCL is characterized by metabolic heterogeneity that is associated with drug resistance and immune microenvironment remodeling [51,52,53]. Understanding the metabolic features of DLBCL patients is vital for addressing the unmet medical need. Our work demonstrated an increased glutamine metabolism in DLBCL patients, consistent with the findings that glutamine metabolism is up-regulated to meet the demands of tumorigenesis [54,55,56]. In addition, the critical metabolite of glutamine metabolism, known as glutamine, was associated with adverse outcomes, indicating the prognostic value of glutamine in DLBCL. Given the limitations of single-center samples in representing the disease characteristics, a large sample validation will be conducted in future studies.
The importance of glutamine metabolism in DLBCL refers to its contributions to the TCA cycle, in which the glutamine metabolite α-KG is used as specific fuel [21]. As a highly aggressive lymphoma, α-KG of DLBCL patients was maintained at a low level, indicating an active energy metabolism in DLBCL cells. Since tumor invasiveness was associated with increased serum LDH (ratio>1) [36], serum α-KG was negatively correlated with serum LDH in DLBCL patients. However, α-KG levels showed no significant changes in different LDH-ratio groups, which may be limited by the insufficiency of patient samples. Recent studies demonstrated that the accumulation of intracellular α-KG leads to tumor-suppressing effects [57]. On one hand, α-KG accumulation disrupts glutamine anaplerosis by reversing glutamine-mediated nitrogen metabolism [54, 58]. On the other hand, α-KG is associated with induction of cell death, including pyroptosis and ferroptosis. Accumulative α-KG resulted in oxidative stress, which affected the redox state of cancer cells and ferroptosis induction [59, 60]. Specifically, α-KG induced ferroptosis by altering the redox state in OCI-LY1 and OCI-LY10 cell lines which harbor c-MYC rearrangement/amplification and BCL-2 rearrangement [61]. MYC hyperactivation could enhance the tolerance to oxidative stress and DNA damage, leading to poor prognosis in DHL patients [38]. Interestingly, α-KG-induced ROS significantly disrupted redox homeostasis, which contributed to overcoming MYC-mediated oxidative stress and oxidative DNA damage. Therefore, α-KG might be considered a promising therapeutic strategy for DHL patients.
As an oxidative, iron-dependent form of non-apoptotic cell death, ferroptosis locates at the intersection of metabolic modulation and signaling transduction [62]. Here, we demonstrated that α-KG-induced ferroptosis relied on lipid peroxidation and TP53-mediated signalings. Previous studies illustrate the critical role of TP53 in predicting outcomes of DLBCL patients [63]. Notably, our work highlighted an important mechanism by which TP53 regulated ferroptosis pathways in DHL cells. Interestingly, differentially activated ferroptosis pathways were detected in DLBCL cells with different TP53 mutation states and genetic features. A previous study illustrated that the steady-state levels of TP53 were higher than that of TP53WT OCI-LY10 cells, even though OCI-LY1 cells were featured with mutant TP53 [64]. Given the stable expression of TP53 in OCI-LY1 cells, α-KG-induced ferroptosis might depend on the TP53-mediated reduction of solute carrier family 7 member 11 (SLC7A11) and GPX4 [65]. In contrast to OCI-LY1 cells, ABC-like OCI-LY10 cells constitutively activated the nuclear factor kappa-B (NF-κB) pathway to resist oxidative stress [66, 67]. Notably, NF-κB activation was associated with iron metabolism, indicating the potential of GPX4-independent ferroptosis in OCI-LY10 cells [68, 69]. Future studies will focus on revealing regulatory mechanisms of ferroptosis pathways in different subtypes of DLBCL.
Another vital function of α-KG refers to the induction of DNA damage. Evidence shows cellular DNA damage regulates cell proliferation and death [70, 71]. In particular, the invasive manifestations and chemoresistance of DHL are associated with the hyperactivation of DDR in DHL cells [38]. Accumulation of intracellular α-KG exacerbated oxidative stress and DNA damage to break the protective effects of DDR and eventually induced cell death. Therefore, our results indicate that α-KG treatment may improve the sensitivity of DDR-targeted therapy in DHL, which is expected to be an effective therapeutic option for DHL patients.
In summary, our study demonstrated the importance of glutamine metabolism in the tumorigenesis of DLBCL and the mechanisms of α-KG in ferroptosis induction. α-KG-based therapy may be a promising strategy for DLBCL, particularly for DHL patients.
Materials and methods
Peripheral serum samples and cell lines
A total of 120 peripheral serum samples, which included 60 patients with initially diagnosed DLBCL and 60 healthy volunteers, were collected from 2020 to 2022 in Shandong Provincial Hospital (Table 1). Serum samples were stored at −80 °C until analysis. The diagnostic criteria were based on the guidelines of the 2017 World Health Organization (WHO) [72]. 53 enrolled patients received treatment, of which 35 patients completed effect evaluation. Therapeutic regimens were shown in Supplementary Table S1. Clinical treatment responses were evaluated after every 3 cycles of treatment, which was based on positron emission tomography/computed tomography (PET/CT) results. Human DLBCL cell lines OCI-LY1, OCI-LY10, Val, and U2932 were grown in Iscove’s modified Dulbecco’s medium (IMDM) (Gibco, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco). Cells were maintained in a 37 °C, 5% CO2 humidified cell incubator. All cells were periodically examined for 15 mycoplasma infection and STR (Short Tandem Repeat).
Untargeted metabolomics
The untargeted metabolomics profiling was performed by Novogene Co., Ltd. (Bei**g, China). Briefly, the peripheral serum samples from 60 DLBCL patients were resuspended with 80% methanol and diluted with LC-MS grade water. After centrifugation, the supernatant was injected into the LC-MS/MS system analysis that consisted of a Vanquish UHPLC system (ThermoFisher, Germany) coupled with an Orbitrap Q ExactiveTM HF-X mass spectrometer (Thermo Fisher). Both positive and negative polarity modes were operated. The raw data files were processed by peak alignment, peak picking, quantitation analysis, and data normalization to predict the molecular formula, followed by the annotation of metabolites. Principal components analysis (PCA) and PLS-DA were performed on the metaX. Differentially expressed metabolites, enrichment, and topology analysis were conducted by the R package.
RNA-seq
To explore the underlying mechanisms of α-KG, the total RNA of DM-αKG-treated OCI-LY1 and OCI-LY10 cells was extracted by TRIzol reagent (Invitrogen, 15596026, CA, USA) and performed RNA-seq experiments (Novogene). Sequencing libraries were generated from purified mRNA by the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, MA, USA) and clustered on the cBot cluster generation system. Subsequently, the prepared library was sequenced on the Illumina HiSeq platform and produced 150 bp paired-end reads. HTSeq v0.6.0 was applied to identify the reads and fragments per kilobase of transcript per million mapped reads (FPKM). GO, KEGG, GSEA, and correlation analysis were performed by using the R package.
ROS detection and apoptosis assay
Intracellular ROS levels and cell apoptosis were measured by flow cytometry analysis. For ROS detection, DLBCL cells resuspended with serum-free IMDM were incubated with 10 mm cell-permeated DCFDA (Abcam, ab113851, MA, USA) for 15 min at 37°C. For apoptosis assay, cells were stained with Annexin V-FITC and PI for 15 min (BD Biosciences, 556547, MA, USA). Fluorescence was then measured by flow cytometry on Navios Flow Cytometer (Beckman Coulter, CA, USA). Images were processed by FlowJo software (v10.8.1, BD, USA).
Lipid peroxidation assay
Cells were harvested and lysed in radioimmunoprecipitation (RIPA) buffer (Beyotime, P0013B, Shanghai, China). The protein lysates were diluted to appropriate concentrations and processed MDA assay using the lipid peroxidation MDA assay kit (Beyotimes, S0131S). The MDA levels were detected by a Multiskan GO microplate reader (Thermo Scientific, Rockford, IL, USA) at 532 nm.
Luciferase-based ATP assay
A luminescent ATP detection assay kit (Abcam, ab113849) was used to measure ATP production. Briefly, cells were lysed and stabilized by detergent solution, further incubating with a reaction mixture containing D-luciferin and firefly luciferase reagent. The luminescence of luciferase activity was recorded by the Ultra-Sensitive Microplate Chemiluminescence (Berthold, LB 960, Germany).
Oxidative DNA damage detection and 2-HG assay
Oxidative DNA damage was measured by an 8-OHdG competitive ELISA kit (Elabsciences, E-EL-0028, Bei**g, China) according to the manufacturer’s protocol. The 2-HG assay was performed as described in the kit protocol (Abcam, ab211070). The optical density of the supernatant was detected by a Multiskan GO microplate reader (Thermo Scientific) at 450 nm.
Neutral comet assay
The neutral comet assay was performed according to the manufacturer’s guidelines (Trevigen, Maryland, USA). DNA fragments were stained by DAPI (Solarbio, S2110, Bei**g, China) and captured by the Olympus BX51 fluorescence microscope. The Open Comet software (Cambridge, USA) was used to analyze tail moments, of which 50 individual cells were calculated in each experiment.
LDH detection assay and morphology observation
Serum LDH levels of the cells were measured by the CytoTox 96 non-radioactive cytotoxicity assay kit (Promega, G1780, Madison, USA). For morphology observation, cells were plated into 6-well plates (1 × 105 cells/well) and observed by OLYMPUS CKX41 inverted microscope with ×200 objective. Each well was observed in 3 different fields and the respective image was processed by Image J software (National Institutes of Health, USA).
Cell proliferation assay and reagents
Cell proliferation rates and viability were measured by the cell counting kit-8 (CCK-8) assay (Do**do, CK04, MD, USA). Cells were seeded in 96-well plates at 1 × 104 cells/well, harvested, and stained with 10 μl CCK-8/well, of which optical density was detected at 450 nm by Multiskan GO Microplate Reader (Thermo Scientific). DM-αKG was from Sigma Aldrich (Cat# 349631, Darmstadt, Germany). Fer-1 was from Selleckchem (S7243, TX, USA). MDH1 inhibitor MDH1-IN-2 was from MCE (HY-147791, New Jersey, USA).
WB
WB was performed as previously described [73]. Cells were lysed in RIPA buffer (Beyotime, P0013B) supplemented with protease inhibitors. 40 μg of total proteins were loaded in the SDS-PAGE (Bio‐Rad, California, USA) and transferred to nitrocellulose membranes. According to the molecular weight of the target protein, the membranes were blocked with 10% skimmed milk and further incubated with primary antibodies at 4°C overnight. The membranes were re-probed with secondary antibodies for 1 h at room temperature. Antibodies were listed in Supplementary Table S2.
In vivo mice models
The animal experiments were conducted by guidelines approved by Shandong University Hospital’s Institutional Animal Care and Research Advisory Committee. Four-week-old female severe combined immunodeficiency (SCID) beige mice of specific pathogen-free (SPF) class were purchased from the Vital River Laboratory Animal Technology Co., Ltd. (Bei**g, China). To construct xenograft DLBCL models, 1 × 107 wild-type OCI-LY1 cells were suspended in 200 μL PBS and subcutaneously injected into the right lower limb of mice. When tumors reached 100 mm3, mice were blindly assigned to two groups (n = 6/group) and injected with either vehicle (PBS, 100 μL per mouse) or DM-αKG (500 mg/kg), administered via intratumoral (i.t.) injections once per day for 5 days. Tumor volume was measured every 2 days and calculated as V = (l × w2) × 0.5. Mice were sacrificed until any one of several criteria were met, which included severe lethargy, more than 10% body-weight loss, and approximately 2 cm of tumor diameter.
Statistical analysis
Each experiment was carried out at least 3 times, every individual experiment containing 3 replicates. A minimum of three independent experiments were displayed with mean ± standard deviation (SD). SPSS Statistics version 20.0 and GraphPad Prism software (v8.0a, La Jolla, CA, USA) were applied to the statistical analysis. The statistical significance between the two groups was determined by the unpaired two-tailed t test with assumed normal distribution. Three or more groups were analyzed by Welch’s one-way ANOVA analysis if normality tests passed, followed by Tukey’s multiple comparison tests. If normality tests failed, the Kruskal-Wallis test was performed with Dunnett’s T3 tests. Proliferation curves were measured by two-way ANOVA analysis with Sidak correction. Significant statistical significance was defined as P value < 0.05. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
