
Abstract
Phenotypic and functional changes in vascular smooth muscle cells (VSMCs) contribute significantly to cardiovascular diseases (CVD) but factors driving early adverse vascular changes are poorly understood. We report on novel and important roles for the Brn-3b/POU4F2 (Brn-3b) transcription factor (TF) in controlling VSMC integrity and function. Brn-3b protein is expressed in mouse aorta with localisation to VSMCs. Male Brn-3b knock-out (KO) aortas displayed extensive remodelling with increased extracellular matrix (ECM) deposition, elastin fibre disruption and small but consistent narrowing/coarctation in the descending aortas. RNA sequencing analysis showed that these effects were linked to deregulation of genes required for calcium (Ca2+) signalling, vascular contractility, sarco-endoplasmic reticulum (S/ER) stress responses and immune function in Brn-3b KO aortas and validation studies confirmed changes in Ca2+ signalling genes linked to increased intracellular Ca2+ and S/ER Ca2+ depletion [e.g. increased, Cacna1d Ca2+ channels; ryanodine receptor 2, (RyR2) and phospholamban (PLN) but reduced ATP2a1, encoding SERCA1 pump] and chaperone proteins, Hspb1, HspA8, DnaJa1 linked to increased S/ER stress, which also contributes to contractile dysfunction. Accordingly, vascular rings from Brn-3b KO aortas displayed attenuated contractility in response to KCl or phenylephrine (PE) while Brn-3b KO-derived VSMC displayed abnormal Ca2+ signalling following ATP stimulation. This data suggests that Brn-3b target genes are necessary to maintain vascular integrity /contractile function and deregulation upon loss of Brn-3b will contribute to contractile dysfunction linked to CVD.
Similar content being viewed by others


Introduction
Cardiovascular diseases (CVD) remain a leading cause of morbidity and mortality, globally [1,2,3] but the chronic nature of the disease means that early changes that contribute to disease development and progression are still not fully understood. In fact, abnormal vasculature such as reduced arterial compliance and blood vessel (BV) contractility are independent predictors of CVD mortality, particularly in patients with essential hypertension [4,5,6]. While vascular damage and dysfunction often develop gradually over time, such changes can remain asymptomatic and undetectable at early stages when interventions may be effective for reducing or preventing adverse changes. At later stages, symptoms arising from vascular damage and arterial stiffening are often irreversible and therefore difficult to treat [2, 7].
Therefore, studying the mechanisms that control integrity and function in healthy blood vessels can provide insight into pathophysiological processes that contribute to vascular dysfunction and disease. Large BVs, such as the aorta provide unique opportunities to study vascular dysfunction because such BV’s are highly adapted for sensing pulsatile blood flow and responding rapidly to changes in cardiac output and blood pressure, under different conditions [8]. In the aorta, differentiated vascular smooth muscle cells (VSMCs) in the tunica media (TM), are critical for rapid adaptation to changes in blood by producing elastin (Eln) that confers elastance and recoil [8, 9]. However, VSMCs also produce contractile proteins [e.g. smooth muscle protein 22-alpha (SM22α), alpha smooth muscle actin (α-SMA) and calponin] that regulate vascular tone [10], blood flow and pressure [11, 12], in response to rapid and active changes in intracellular calcium (Ca2+), which is, in turn, regulated by the activation or inhibition of Ca2+ channels, pumps and other regulatory proteins found in the plasma membrane and intracellular store e.g. sarco-endoplasmic reticulin (S/ER) [13,14,15]. Therefore, controlling the genes that encode such essential proteins in VSMCs, will be critical for determining vascular function.
This is particularly important because of the inherent plasticity of VSMCs, whereby contractile cells can undergo phenotypic switching in response to injury or chronic stress [e.g. raised intracellular Ca2+ or S/ER Ca2+depletion and stress; increased reactive oxygen species (ROS), inflammation [16,17,18,19,20]. Under such conditions, VSMCs undergo de-differentiation and switch from contractile cells into mesenchymal phenotypes that acquire migratory, proliferative and secretory properties [9, 14]. Such effects are driven by changes in genes that encode important cellular proteins such as reduction in contractile proteins but increase expression of extracellular matrix (ECM) proteins e.g. collagen that reduces VSMC contractility and contributes to vascular dysfunction such as arterial stiffening and hypertension [12, 14] highlighted.
Transcription factors (TFs) that regulate the rate of transcription of multiple target genes, in a tissue specific manner are critical for determining cell fate [21] and changes in key TFs required for normal vascular integrity and function can lead to adverse vascular changes linked to CVD [22]. The Brn-3b/POUF2 protein (Brn-3b) TF, was identified as a key regulator of gene expression in cardiovascular tissues [23,24,25,26]. This TF is characterised by a highly conserved DNA binding POU (pit-oct-unc) homeodomain that can bind to unique BRNF DNA sites in specific gene promoters and either activate or repress gene transcription by RNA polymerase II, depending on cell/tissue type [27,28,29]. However, Brn-3b can also indirectly regulate gene transcription by binding to and modulating the transcriptional effects of other TFs e.g. p53 tumour suppressor protein and oestrogen receptor (ER) on distinct target genes [24, 25, 28, 30]. Interestingly, the gene encoding Brn-3b consisting of 2 exons and can give rise to 2 distinct isoforms, thought to arise from alternative promoter usage, with the longer Brn-3b (l) protein (43–46 kDa) encoded by exon 1 and exon 2 while the shorter Brn-3b(s) isoform encoded entirely by exon 2 [30,31,32]. While the precise function of the different Brn-3b isoforms remain unclear, experimental evidence support auto-regulation and positive feedback loop whereby different isoforms can be regulated by each other depending on the tissue of expression [30,31,32,33].
Although isolated and widely studied in neuronal cells, Brn-3b is also found in other tissues including cardiovascular system (cardiomyocytes/heart), metabolic tissue (skeletal muscle; adipose tissue), testes and immune cells [peripheral blood mononuclear cells (PBMC), T-cells and monocytes)] [30, 33, 34]. Importantly, Brn-3b target genes control many essential cellular processes including proliferation, metabolism, differentiation and apoptosis, depending on the cell type and growth conditions [24, 26, 30, 35]. For instance, Brn-3b controls survival and differentiation in retinal ganglion cells by regulating sonic hedgehog, myostatin (Gdf8) and Pax4 [36, 37]. However, in epithelial-derived tumour cells, Brn-3b promotes cell proliferation by activating cell cycle proteins, cyclinD1 and CDK4 while repressing the Brca1 tumour suppressor gene [38, 39]. Yet in metabolic tissues including skeletal muscle and adipose tissues, Brn-3b is implicated in regulation of glucose homoeostasis by controlling expression of Glut4 and GSK3β, since reduction or loss of Brn-3b is linked to hyperglycaemia and insulin resistance [35].
Brn-3b is highly expressed in foetal hearts and while expressed at low levels in normal adult hearts, this TF is increased in adult hearts following acute injury (e.g. coronary artery ligation) [24] or in response to chronic stress e.g. angiotensin II treatment, which induces ventricular hypertrophy [26]. Recent studies using male Brn-3b KO mutant mice have shown that loss of Brn-3b caused subtle differences in contractility and cardiovascular function at baseline e.g. reduced arterial elastance and end systolic pressure volume relationship [26]. Moreover, Brn-3b appears to be necessary for normal adaptive responses in male hearts because angiotensin II treatment in Brn-3bKO mutants caused extensive fibrosis in the ventricular wall and remodelling around the coronary vasculature, which was linked to adverse functional responses including reduced cardiac output and ejection fraction [26].
Remodelling around Brn-3b KO coronary arteries pointed to potential roles for this TF in controlling vascular integrity and function(35) especially since published genome wide association studies (GWAS) data have identified SNPs associated with coronary heart disease (CHD), coronary artery diseases (CAD) and cerebrovascular accident (CVA) within the genomic region chromosome 4q (31.2), containing the Brn-3b genomic locus [40,41,42,43,44,45]. However, to our knowledge, the expression and effects of Brn-3b in the vasculature are still unknown.
In this study, we confirmed Brn-3b expression in the aorta, with localisation to VSMCs. Studies using Brn-3b KO mice showed that loss of this TF caused significant histological and structural changes including small but consistent narrowing of the aorta, thickening of the tunica adventitia (TA) and disrupted Eln fibres in aortas from male mice. This was associated with increased wall stiffness and attenuated contractile responses to KCl, PE and U46619. RNA sequencing analysis and subsequent validation studies showed that loss of Brn-3b affected genes such as Cacna1d; S/ER Ca2+ channel, rayanodine receptor RyR2, Pln and reduced Atp2a1, which are implicated in controlling Ca2+ signalling and primary VSMCs cultured from Brn-3b KO aortas displayed abnormal Ca2+ responses to ATP. Similarly, reduced chaperone genes e.g. Hsph1, Hspb1, DnajA1 in Brn-3b KO aortas, which is linked to increased ER stress [20, 46] was associated with phenotypic switching and increased proliferation in Brn-3b KO VSMC cultures. Taken together, these results suggest that Brn-3b TF controls genes that are required for maintaining calcium homoeostasis, contractility and stress responses in VSMCs and loss of Brn-3b will lead to vascular dysfunction.
Materials and methods
Materials
General laboratory reagents: Merck (Nottingham, UK) and Sigma (Dorset, UK), unless otherwise stated. Primary antibodies: Brn3b-rabbit pAb (ab128849- now discontinued). Abcam-Cambridge, UK); Brn3b-rabbit pAb Biorbyt ORB576708; orb 1313 (now discontinued); GAPDH and β-tubulin (Cell Signalling Technology, USA) α-SMA (Ab7817;Abcam-Cambridge, UK); Col1a1 (ab88147Abcam, Cambridge, UK); Ki67- M7249 (Dako, Cambridgeshire, UK). HRP-conjugated secondary Ab, Dako (Cambridgeshire, UK).
qRT-PCR Primer Sequences
Brn-3b F - 5′ GAGAGAGCGCTCACAATTCC 3′;
Brn3b R- 5′ ATGGTGGTGGTGGCTCTTAC 3′
36b4 F- 5′ AGATGCAGCAGATCCGCAT 3′;
36b4 R- 5′ GTTCTTGCCCATCAGCACC 3′
Gapdh F- 5′ CTTCATTGACCTCAACTAC 3′;
Gapdh R 5′ AGTGATGGCATGGACTGTG 3′
Ryr1 F- 5′ GCAGGAGACGTACAGTCAGG 3′;
Ryr1 R – 5′ CAAGGATGTCTGCACGGAGT 3′
Ryr2 F-5′CCTACAGTGGTATGTATCTTTGCTGTCT3′;
Ryr2 R- 5′CTCTTGAAGGCCAACATC GAA 3′
Cacna1d- F-5′CGTGCCTTCCGAGTGTTAAG3′;
Cacna1d- R-5′GGACCATGGCTTTTATAATGGA 3′
Cacnb2 F- 5′TGGAGTCGACTTTTTGCCGA 3′
Cacnb2 R- 5′ TCCATAGGACTGTGCTCCGA 3′
Pln F- 5′ ACCGAAGCCAAGGTCTCCTA 3′;
Pln R- 5′ TAGCCGAGCGAGTGAGGTAT 3′
DnaJa1 F- 5′ CCGCTCACCGGCTGTAAA 3′;
DnaJa1 R- 5′ GGGTGGTACTTCAAGGCCAA 3′
Hspa8 F- 5′ CTGCTGCTATTGCTTACGGC 3′;
Hspa8 R- 5′ TCAAAAGTGCCACCTCCCAA 3′
Hsph1 F- 5′AACCCCAGATGCTGACAAAG 3′;
Hsph1 R- 5′GCAGCTCAACATTTACCACCT 3′
Atp2A1 F- 5′AAGGCTCGGGACATCGTT 3′;
Atp2A1 R- 5′GGATGTCTGCAGGGACTTTG 3′
ELNF1 F- 5′ GCTGATCCTCTTGCTCAACC
ELNF1 R- 5′ CAATACCAGCCCCTGGATAA
MMP-2 F- 5′TGCCCCCATGAAGCCTTGT3′
MMP-2 F- 5′TACAGCTGTTGTAGGAGGTGCC 3′
Methods
Experimental models
All animal experiments were carried out in compliance with UK Home Office regulations (Animals Scientific Procedures Act 1986) and approved by local UCL Ethics Review Board. Early studies used Wistar rats or wild-type (WT) C57Bl/6 mouse models purchased from commercial companies (Harlan UK). Later studies with knockout (KO) mice and WT littermate controls were obtained by crossing Brn-3b heterozygotes C57Bl/6 strains. For all experiments involving animal models, a priori sample power calculations were carried out by using multiple endpoints with online tools e.g. IACUC power calculator, to determine the sample sizes required to achieve statistical significant (e.g. 80% power and 95% confidence limits) to detect prespecified effects. Randomisation studies and blinding during analysis were not possible for these experiments because of the requirement for consistency in processing diverse data sets with different endpoints.
Histological staining
To analyse for morphological and structural changes in the aortic sections, histological staining was undertaken using Haematoxylin and Eosin (H&E) staining; Masson′s trichrome staining (Abcam-Cambridge, UK) or oil red O staining (Sigma, UK). For these studies, formalin-fixed, paraffin-embedded aortic sections from age matched Brn-3b KO and WT mice, were dewaxed, rehydrated and stained according to the manufacturer’s protocol. To quantify TA thickness matched aortic sections were analysed using NDPview-2 software (Hamamatsu, Japan), by undertaking ~50 equidistant measurements of the aortic wall. Data represents mean ± SD from ≥6 matched, independent WT or Brn-3b KO aortic sections.
Protein preparation and quantification
Total protein extracts were prepared either from mouse aortas or primary VSMC cultures as follows. For aortic samples, intact aortas dissected from 2 to 3 mice in each group (WT controls or Brn-3b KO mutants) were processed using standard protocols [26] i.e. dissociated snap frozen tissues resuspended and processed in RIPA buffer. Total protein content was quantified using BCA (bicinchoninic acid) kit (Merck, UK) and ~30 μg/well protein extracts were resolved on SDS-PAGE gels then transferred onto PVDF membranes overnight [24]. Following incubation (1 h at RT) with block buffer [4%milk + phosphate buffered saline + 0.1% Tween-20 (PBST)], and primary antibody for 2 h at RT or overnight at 4 °C (optimised for specific antibodies); membranes were washed (X5) then incubated with HRP-conjugated 2nd antibody (1 h; RT). Signals were developed using enhanced chemiluminescence (BioRad, UK) on Syngene G:BOX imager and protein quantification done out using FiJi (ImageJ) software. Variation in protein loading was adjusted with β-tubulin (housekee** protein).
Co-immunostaining using colorimetric or fluorescent detection
Immunostaining for protein localisation was done using paraffin-embedded tissue sections or cultured cells. Tissue sections were dewaxed and rehydrated prior to antigen retrieval (0.01 M sodium citrate pH6.0, 10 min), and single or double immunostaining protocols undertaken using Vectastain Elite ABC Kit (Vectorlabs) or ImmPRESS® Duet Double-Staining Kit, (according to the manufacturer’s protocol [25, 26]). Primary antibodies were incubated in a humidified chamber, either 2 h (RT) or overnight (4 °C) and control samples were incubated with second antibody only. The manufacturers protocol was modified to include additional washes (5 PBST; 5 min) after which detection was undertaken using Vectastain Elite ABC Kit (Vectorlabs) or ImmPACT® DAB EqV Substrate (HRP, brown); ImmPACT Vector® Red Substrate (AP, magenta). Following graded ethanol dehydration, cover-slipped slides were imaged using the Hamamatsu Nanozoomer (Hamamatsu, Japan) then analysed using NDP view 2 software.
Fluorescent immunostaining of cultured cells was undertaken as previously described [24]. Briefly cells were fixed in 4% PFA and permeable if necessary. Following incubation with block solution (20% goat serum in PBST) antibodies were incubated either overnight at 4 °C or 2 h at room temperature. Following 5× washes in PBST and incubation with appropriate fluorescent tagged, secondary Ab, cells were imaged using fluorescent microscopy and Leica software for analysis (Leica DMi8, LAS X).
Transmission electron microscopy
Transmission electron microscopy (TEM) were undertaken using resin embedded sections prepared from Brn-3b KO mutant aortas and wild-type controls using well established methods. Briefly, aortas were perfused and fixed in 100 mM cacodylate buffer containing 2.5% glutaraldehyde overnight at 4 °C. Samples were transferred to 1% osmium tetroxide, dehydrated in an ethanol series, and then embedded in araldite resin (Electron Microscopy Sciences, Hatsfield, PA). 0.5 μm sections were stained with toluidine-blue and analyses using light microscopy. For electron microscopy, ultra-thin sections (70–80 nm) were cut and set on 200-mesh copper. The grids were counterstained with lead citrate for 3 min and imaged on a Jeol 1400 electron microscope with a Rio Gatan camera.
RNA sequencing and data analysis
For RNA sequencing, total RNA was prepared from aortas taken from Brn-3b KO male mutants and age-matched wild-type controls (>3/set). Quality controls (QC) was done using NanoDrop and Agilent Bioanalyser 2100 before library preparation was carried out using KAPA mRNA HyperPrep Kit (p/n KK8580) according to manufacturer’s instructions. During this process, highly purified mRNA, fragmented by chemical hydrolysis and primed with random hexamers, was used for RNA-dependent cDNA synthesis with “A-tailed” at 3′ end, to prevent self-ligation and adaptor dimerisation. Amplification and first strand library preparation was undertaken using adaptors for PCR with high fidelity polymerase and libraries were sequenced on the NextSeq 500 instrument (Illumina, San Diego, US) using either a 43 bp or 81 bp paired end run with general yield of ~15 million reads per sample. De-multiplexed data converted to Fastq files (Illumina’s bcl2fastq Conversion Software v2.19) were used for alignment with the reference genome using STAR (v2.5b) and aligned data were deduplicated (Picard Tools v2.7.1) and reads per transcript counted by FeatureCounts (v1.4.6p5). Ensembl ID for each gene was determined using g:Profiler and this data was then used for normalisation, modelling and differential expression analysis using the iDEP.93 software platform, which connects multiple R/Bioconductor packages with annotation and pathway databases to provide comprehensive analysis of RNA sequencing data
Data analysis using iDEP.93 platform
Preprocessing was done to filter genes with ≥0.5 counts per million in at least one sample inclusion in the analysis using EdgeR (default settings CPM-0.5, pseudo-count c-4) and principal component analysis was undertaken using PC1 and PC 3 to visualise the relationship between sample groups based on sample variance. For K-means clustering analysis, read counts were transformed to log2 and the 2500 most variable genes (based on standard deviation ranking) were separated into one of four clusters, based on functionality, molecular and biological processes with students t-test used to calculate the values for enriched pathways within each cluster. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis provided a systematic analysis of pathways associated with genes in each cluster (adjusted p value). Similarly, Gene Ontology (GO) database platforms provided information on the association of genes within each cluster with specific biological processes, molecular function and cellular components. The Disease.Jensen.DISEASES database also linked to this platform, was used to identify diseases associated with specific subsets of genes, depending on adjusted p values. Gene Set Enrichment Analysis (GSEA) was undertaken to identify genes that were either up- or down-regulated in Brn-3b KO vs WT, using the DESeq2 method, which calculated the p values using the Wald test and used the read count data (unlike K-Means Clustering which uses transformed data). Analyses were undertaken using absolute values of fold changes in the pre-ranked mode (preranked fgsea) and were based on comparison between genotypes i.e. KO vs WT, with 15–2000 gene set (min; max), pathway significance cut-off of 0.1 false discovery rate (FDR) and minimum fold change of 1.5, with top 30 pathways selected. Normalised enrichment score (NES) accounted for gene set sizes and correlations between gene set and expression data, with positive and negative enrichment scores (ES) indicating correlation with Brn-3b KO and WT phenotypes respectively. KEGG and GO pathway analyses were used to determine functional changes associated with up- or down-regulated gene sets.
Differential Expression of Genes (DEG) was also carried out using DESeq2 method to identify genes with significant and concordant differences in a-priori gene sets in the different variables (Brn-3b KO vs WT). The minimum fold change was set at 1.5 and with 0.10 FDR cut off. WT values were used as the control baseline for the analyses. Functionality of up and/or downregulated was determined using g:Profiler.
Primary VSMC cultures
Primary VSMC cultures were prepared from freshly dissected Brn-3b KO and WT aortas as described [47]. Briefly, excess perivascular tissue and tunica adventitia were carefully removed from isolated aortas (to prevent contamination with fibroblast of perivascular adipose tissue (PVAT)) and ~2 mm of cleaned aortic rings were carefully placed into a T25 flask, in large drops of growth medium (DMEM-F12 + 1% Penicillin-Streptomycin +15%foetal calf serum) and left to attach to the flask. After 5–10 min, culture medium was gently added to cover flask, which was then placed in a humidified incubator (37 °C and 5% CO2). Media were changed every 2–3 days until cells were confluent and ready for experiments (up to 10–14 days).
Gene validation: RNA extraction, cDNA synthesis and quantitative polymerase chain reaction (qRT PCR)
RNA was extracted from snap-frozen aortas taken from Brn-3b KO and WT aortas. Tissues were crushed in liquid nitrogen and homogenised in RLT buffer then RNA was extracted using Qiagen RNeasy mini kit (Qiagen, Manchester, UK) or TRIZOL® Reagent (Invitrogen). Since Brn-3b(s) is only encoded by exon 2, it was not possible to use primers spanning the intron. As such, it was necessary to remove all contaminating genomic DNA prior to cDNA synthesis, which was achieved using RNAse-free DNAse1 (Promega, Southampton, UK). 1 μg of total RNA from each sample was used for cDNA synthesis, using Superscript™ II Reverse Transcriptase (Invitrogen, UK), according to the manufacturer’s protocol.
Validation studies to confirm changes in RNA sequencing data for selected genes was undertaken using q-RT-PCR with Eppendorf Mastercycler using SYBR Green chemistry. Primers designed for each target gene were used to amplify cDNA for specify genes using independent WT or KO aortic RNA. Housekee** genes e.g. GAPDH and 36B4 were used correct for RNA variability between samples and a reference sample was included to facilitate use of ΔΔCT method to calculate fold changes in relation to controls samples. Statistical analysis was undertaken using results from multiple experiments using independent aortas with student’s t test used to show significance, *p ≤ 0.05.
Wire myography for contractility studies in Brn-3b KO and WT aortic vascular rings
Thoracic aorta from Brn-3b KO and WT mice were prepared by removing perivascular vascular tissue. The cleaned aortae were used to generate vascular rings for wire myography during which tissues were bathed in Krebs solution at 37 °C with a 95% O2 and 5% CO2 environment. Vascular rings were mounted on the myograph (Danish Myo Technology, Hinnerup, Denmark) and DMT normalisation module used for isometric tension recordings. At baseline, the optimal pre-tension condition established by stretching to 95% internal circumference with resting transmural pressure of 100 mmHg (IC100). Following equilibration and normalisation of vascular reactivity, constricting cumulative dose-responses were measured following treatment with potassium chloride (KCl, 7.50–90 mM/L), phenylephrine (PE, 100 nM/L to 100 μM/L) (Sigma-Aldrich, Missouri, USA) and U-46619 (0.1–25 nM/L) (Cambridge Biosciences, Cambridge, UK). Results shown at raw force (mN) [48, 49].
Calcium imaging using VSMC
For these studies, primary VSMC cultures derived from single WT and Brn-3b KO mouse aortas were plated into glass FluoroDish petri dishes (WPI World Precision Instrument, Thermo Fisher Scientific, UK) and grown to ~70% confluency. Prior to signal imaging and analysis, cells were transferred to calcium free growth medium then loaded with fluo-4-AM dye (Thermofisher Scientific) by adding to HBSS in presence of 0.02% pluronic F-127 and incubating at 37 °C in 5% CO2 for 30 min, according to manufacture instructions in. Cells were washed twice in HBSS and maintained in 900 uL of HBSS at 37 °C in 5% CO2. Basal fluorescence signals were measured and recorded using the Zeiss LSM 880 Observer Z1, Airyscan confocal microscope after which, ATP was carefully added to the dish to a final concentration of 100 uM and changes in fluorescence intensity were recorded for 400 at 1 frame per sec. Since each sample was analysed individually, measurements were done by alternating WT and KO for pairwise comparison. Time-lapse movies were analysed using ImageJ with >10 cells analysed for each sample. For each cell, the changes in signals over time were expressed as fold change of the fluorescence at baseline and for each VSMC culture, a minimum of ten cells were analysed to obtain the average signals for that sample. The final data represents data from six independent studies carried out in individual aortas.
Analysis of VSMC proliferation rates using BrdU incorporation studies
The rate of proliferation was measured by determining DNA synthesis using 5-bromo-2- deoxyuridine (BrdU) Cell Proliferation ELISA Kit (Abcam, Cambridge, UK) and was carried out according to the manufacturer’s protocol. Briefly, sub confluent VSMC cultures from Brn-3b KO or WT aortas were pulse labelled with BrdU for 2 h. Cells were fixed for 30 min then washed prior to addition of the primary detector antibody for 1 h at room temperature (RT). Following washes, peroxidase conjugated second antibody was incubated (30 min RT). Following incubation with the peroxidase substrate (30 min, RT), the reaction was stopped and signals were quantified using spectrophotometer microtitre plate reader set at dual wavelength (450/550 nm).
Statistical analysis
Statistical analysis was carried out using GraphPad Prism or Excel. To determine statistical significance within different dataset analysis were undertaken using T-test, F-test or 2 way ANOVA with Bonferroni post-test. T-test was used to determine p value and standard deviation while F test were used to compare variances and determine significance of differences. Two-way ANOVA also provided detailed comparisons between groups with additional data including sum-of-squares main square and analysis of sources of variation.
Results
Brn-3b expression and localisation in the aorta and primary VSMC cultures
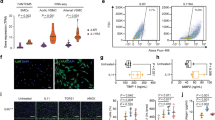
To investigate expression and roles for Brn-3b in BV, pooled aortas from wild-type (WT) mice were used to analyse for protein expression using western blot (WB) analysis. Fig. 1a shows that Brn-3b(s) protein isoform was readily detectable in pooled aortic protein extracts from WT mice but not in Brn-3b KO aortic extracts while β-tubulin blot is included to show variability in total cellular protein. It should be noted that only the shorter Brn-3b(s) isoform was detected in aortic tissue extracts (full blot in Supplementary Fig. 1 also shows positive control, testis, primarily expressing the Brn-3b(l) isoform).
Brn-3b protein localisation in aortic sections was also analysed by undertaking co-immunostaining studies with α-SMA antibody used to mark VSMCs. Studies were carried out using aortic sections from WT and Brn-3b KO aortas. As shown in Fig. 1b, Brn-3b protein (brown stain) was detected mainly to the medial layer in WT aortic sections, where it was co-localised with the VSMC marker, α-SMA (magenta) but not in control WT sections stained with secondary Ab only.

a WB showing expression of the shorter Brn-3b(l) protein in aortic extracts prepared from pooled WT aortas (dissected from 2–3 mice) compared with pooled aortas from Brn-3b KO mutants (negative control). β−tubulin blots indicate variation in protein loading. b Representative images of immunostained WT aortic sections showing co-localisation of Brn-3b protein (brown stain) with VSMC marker, αSMA (magenta). Negative control = 2nd Ab and α-SMA. Different layers of the aorta: TI tunica intima, TM tunica media, TA tunica adventitia. c quantification of Brn-3b mRNA in primary VSMC cultures prepared using aortas from WT mutant n-3b KO mice (n = 5 independent cultures). Analyses were carried out using student’s t-test. d Representative immunofluorescent images of aortic VSMCs from WT mice (top panel) or Brn-3b KO mice, co-stained with Brn-3b (green) and α−SMA (red). DAPI mount indicates cell nuclei (blue).
To confirm Brn-3b expression in VSMCs, primary cultures were isolated and grown from WT or Brn-3b KO mouse aortas and either used for quantification (qRT-PCR) or localisation studies (co-immunostaining). qRT-PCR data shows that Brn-3b mRNA was detectable in VSMC cultures prepared from WT but not Brn-3b KO aortas (Fig. 1c). Similarly, co-immunostaining studies confirmed Brn-3b antibody staining (green) colocalized with α-SMA (red) in VSMC derived from WT aortas (top panel). Comparison with DAPI staining (blue) showing cell nuclei indicated that Brn-3b was expressed in the nuclear and perinuclear regions in aortic VSMCs. As expected, Brn-3b was undetectable in VSMC from Brn-3b KO mutant aortas (bottom panel) in cells that express α-SMA (red).
Abnormal structural and histological changes in Brn-3b KO aorta
Since loss of Brn-3b was linked to extensive remodelling around the coronary arteries in male Brn-3b KO hearts [26], we next analysed for structural changes in aortas from male Brn-3b KO mice, when compared with WT controls. Fig. 2a shows that aortas from Brn-3b KO mice displayed small but consistent narrowing/coarctation in the descending aortas, when compared with age and gender-matched WT controls. H&E staining of aortic sections also showed marked histological changes in Brn-3b KO aortas (Fig. 2b), which included thickening of the adventitial (TA) layer and increased white adipose tissue (WAT) deposits in PVAT, when compared with WT aortas. Such changes were also confirmed using Masson’s trichrome staining, with Fig. 2 showing increased deposition of ECM proteins (blue) in the thickened adventitial layer (TA) of Brn-3b KO aortas (ii), when compared with WT aortas (i). These differences were also confirmed by quantifying changes in TA thickness and lumen diameter in transverse aortic cross-sections from independent age-and gender matched WT or Brn-3b KO aortas (n > 6). Figure 2d(i) shows that Brn-3b KO aortas displayed significant increases in TA thickness but reduction lumen diameter, when compared with WT aortas [Fig. 2d(ii)]. Immunostaining of aortic sections using collagen 1A1 (Col1a1) antibody also confirmed increased Col1a1 staining in the TA layer of Brn-3b KO aortas, when compared with WT controls (Fig. 2e).

a Representative images of intact aortas taken from WT or Brn-3b KO mice, after staining for oil red. Images shown at X5 magnification with boxed areas expanded to highlight narrowing/constriction in Brn-3b KO aortas and corresponding WT aortas. b H&E staining of longitudinal aortic sections from WT (i) or Brn-3b KO mice (ii). 20× magnification of selected areas shows increased adventitial thickness highlighted by dotted lines (arrows indicate specific areas of thickening in KO aortas; * highlights increased WAT deposition) in Brn-3b KO but not WT aortic sections. L lumen. Images (b–g) were captured using Hammamatsu Nanozoomer imaging system and shown at ×5; ×20 and ×40 magnification. TI tunica intima, TM tunica media, TA tunica adventitia, WAT white adipose tissue, PVAT perivascular adipose tissue. c Representative images showing Masson’s trichrome staining of longitudinal aortic sections from either (i) WT or (ii) Brn-3b KO mice which highlights the increases in thickness of tunica adventitia (TA) layer and elastin disruption in tunica media (TM) of Brn-3b KO aortas (*), when compared with WT controls. Red staining represents cytoplasm; dark purple/black shows cell nuclei; blue staining indicates ECM protein (e.g. collagen) deposits. d Representative images showing immunostaining for collagen protein, Col1a1, in WT and Brn-3b KO aorta sections using DAB immunostaining protocol, with intense brown staining indicating increased expression in aortic sections from Brn-3b KO mice compared with WT controls. e (i) Quantification of TA width thickness in representative aortic sections, with each point representing the mean values from eight independent mice within each group. Data represents mean and standard deviation (±SD) with significance (***p < 0.001), determined by students t test. (ii) Graph showing differences in aortic lumen diameter from WT or Brn-3b mice (n = 8 independent aortas/group). Analysis done using students t-test. f Masson’s trichrome stained aortic section highlighting disruption of elastin fibres in Brn-3b KO mutants; indicated by fragmented blue staining (*), compared with WT aortas. g van Gieson staining of elastin fibres (dark brown) in aortic section from WT or Brn-3b KO mice showing disruption of elastin fibres in Brn-3b KO aortas (arrowheads) when compared with WT aortas. Magnification is shown at 5× (top panels) and 40× magnification (bottom). h–j Transmission electron microscopy images showing differences between aortas taken from WT and Brn-3b KO mice. h Increased collagen fibres in the Tunica adventitial (TA) layer with Brn-3b KO aortas showing significantly thicker TA with more compact arrangement of collagen fibres when compared with WT controls. i Representative images of the tunica media (TA) showing elastin fibres and VSMCs found between the elastic fibres (E) are indicated. Disruption of elastin fibres in Brn-3b KO aortas are indicated by yellow arrows, when compared with WT aortas. j Images showing aortic perivascular adipose tissue (PVAT) surrounding the aorta taken from either WT or Brn-3b KO mice. Black arrows indicate larger numbers of infiltrating lymphocytes in Brn-3b KO PVAT when compared with WT controls.
Interestingly, analysis of trichrome staining aortic sections at high magnification also showed significant changes in elastin fibres in Brn-3b KO aortas when compared with WT (Fig. 2f). Therefore, van Gieson staining was carried out on aortic sections to analyse for changes in elastin fibres. Fig. 2g confirmed that Brn-3b KO aortas displayed disrupted elastin fibres compared with WT controls.
Such significant morphological and structural changes in Brn-3b KO aortas were also confirmed at the ultrastructural level, using images from TEM (Fig. 2h, i). Fig. 2h shows increased density and compaction of collagen fibres in the thicker TA layer of Brn-3b KO aortas compared with WT aorta. Similarly, disruption in the elastin lamina in mutant aorta can be clearly seen in Fig. 2i while Fig. 2j shows differences in fat cells but also highlights infiltration of immune cells (arrows) in aortic PVAT from Brn-3b KO aortic sections that was not evident in PVAT from WT aortas. These results confirmed that structural and histological changes linked to morphological abnormalities in Brn-3b KO aortas were associated with ultrastructural defects that could profoundly alter aortic structure and function.
Contractile changes in Brn-3b KO aorta
To determine how structural changes may affect aortic blood flow in Brn-3b KO aortas, echocardiography was undertaken using Visualsonics Vivo 2100 to analyse ascending aortic velocity and aortic root diameter in male Brn-3b KO mutants and age-matched WT controls. Figure 3a shows that ascending aortic velocity was significantly increased in Brn-3b KO mutants, when compared with WT controls, despite no significant differences in aortic root diameter (Fig. 3b), which suggested that loss of Brn-3b may contribute to changes in aortic contractility.

Echocardiography data showing changes in ascending aortic velocity (a) and measurement of aortic root diameter in male Brn-3b KO mice (b), when compared with age-matched WT controls. c Graph showing IC100 measurement (from wire myography) to measure differences in elastic properties of vascular rings from WT and Brn-3b KO mice at baseline. d Summary of data showing responses of thoracic aortic vascular rings from Brn-3b KO aortas and WT controls, following treatment with cumulative concentration of (i) KCl, (ii) PE and (iii) PGH2 prostaglandin analogue, u46619. Graphs show changes in force of contraction (DmN) elicited following different treatments, as shown. Data represents mean and standard error of measurements from vascular rings taken from 6 independent Brn-3b KO and WT aortas. Statistical significance was determined using two-way ANOVA analysis.
Therefore, wire myography was next used to analyse for differences in elastic properties and contractile responses in aortic rings from Brn-3b KO mutants and WT controls using. For these studies, baseline wall stiffness in aortic rings was established using the normalisation method whereby vascular rings were pre-stretched to 95% of internal circumference with resting transmural pressure of 100 mmHg (IC 100). As shown in Fig. 3c, the IC100 values were significantly lower in Brn-3b KO vascular rings which may be linked to changes in lumen diameter (Fig. 2d(ii) but could also reflect differences in the elastic properties of mutant blood vessels.
We next analysed for changes in contractile responses of pre-stretched, equilibrated vascular rings from Brn-3b KO and WT aortas by adding cumulative dose of known mediators of vascular contraction including phenylephrine (PE), prostaglandin analogue, U46619, or potassium chloride (KCl) [50,51,52]. Pooled data from independent aortas, taken from six WT and six Brn-3b KO mice, (Fig. 3d) showed that, as expected, vascular rings from WT aortas displayed dose-responsive increases in force of contraction following treatment (i), with PE causing the most significant responses. In contrast, contractile responses in Brn-3b KO aortas were considerably blunted following PE treatment, indicating that mutant aortas were unable to contract significantly. Similarly, treatment with U46619 (ii), and KCl (iii) also induced strong contraction of vascular rings from WT while responses in Brn-3b KO aortas remained significantly attenuated. These highly reproducible results from independent aortas suggests that pre-constriction of mutant aortas upon loss of Brn-3b may prevent subsequent contractile responses to PE, U46619 or KCl.
RNA sequencing analysis to identify genes altered in Brn-3b KO aortas
To understand the molecular basis for such changes in Brn-3b KO aortas, RNA sequencing analysis was undertaken to identify genes that were differentially regulated upon loss of Brn-3b in the aorta. Following in-depth analysis of data using iDEP.91 software [53], principal component analysis identified one WT outlier (Fig. 4a) but K-means analysis of the top 2500 differentially regulated genes showed that the same gene clusters were identified when using all datasets (Supplementary S-Fig. 1) or when the outlier was excluded (Fig. 4b). However, the outlier caused some skewing in subsequent analyses to identify up- and down regulated genes so was omitted for later analyses with selected genes validated in multiple, independent mRNA from WT or Brn-3b KO aortas.

a Principal Component Analysis (PCA) of all data obtained from RNA sequencing of WT and Brn-3b KO aortas. Samples from all three KO aortas (orange dots) were clustered together (boxed area) but only two WT samples (oval) showed similar patters while the WT sample 2 M (circled), was identified as an outlier with significant differences in both PC1 and PC3 components and was excluded from analyses. b Heat map showing the four clusters arising from K-means clustering of the top 2500 from two WT and three KO samples (excluding outlier sample, 2 M WT). KEGG pathways associated with enriched genes within each cluster are shown i.e. Red = upregulated genes; green = down regulated genes.
The most consistent and significant gene expression changes between WT and mutant aortas were observed in 2 clusters, A and C (Fig. 4 and Supplementary Fig. 1b; Supplementary Table 1b). KEGG pathway analysis showed that genes in cluster A (844 genes; adjusted p values < 8.0e-0.3) were strongly associated with calcium signalling (adjP 3.4e-05) and muscle contraction (adjP 3.4e-05) (Fig. 4) that directly affect vascular function while GO analysis confirmed that genes within this cluster were mainly associated with the plasma membrane or ion channels in the S/ER and participated in biological processes including sarcomeric structures and ion transport, (Supplementary Table 1c, d). Since Ca2+ signalling genes are also implicated in controlling cardiac contractility and function [54,55,56], it is unsurprising that cardiac pathways including adrenergic signalling, cAMP signalling were also identified (s-Table 1e). Interestingly, other affected genes within this cluster were linked to regulation of circadian entrainment (adjP 1.8e-03) and metabolic processes (adjP 5.3e-03), which can indirectly contribute to vascular dysfunction [57,58,59] (Supplementary Table 1). In contrast, genes in cluster C (521 genes; adjusted p values < 2.6 e-0.3) were mainly linked to regulation of VSMC contraction (adjP 3.6e-03) but also immune responses (adjP 2.6e-03), which can indirectly alter vascular function (Supplementary Table 1a) [12, 60, 61]. GO analysis also confirmed that cluster C genes were implicated in VSMC contraction and muscle development/differentiation. Affected genes in cluster B (617 genes; adj p values < 7.5e-0.3) and D (542 genes; adj p values < 5.3e-0.3), were linked to haematopoietic cell lineage and immune responses respectively but the sample-to-sample variation limited any meaningful interpretation of this data.
Identification of up- and down-regulated genes
GSEA was next combined with KEGG pathway and GO analyses to identify how genes that were specifically up- or down-regulated in Brn-3b KO aortas affected key functional pathways that were linked to essential biological processes. Table 1a shows the largest number of upregulated genes in Brn-3b KO aortas (158 genes) were implicated in Ca2+ signalling and related pathways (cAMP signalling and β-adrenergic signalling) with significant normalisation enrichment scores (NES+ 1.51) while 93 genes (NES + 1.45) were implicated in VSMC contraction. Supplementary Table 1 (s-1) shows the list of genes within each cluster. In support of essential findings for such differential gene expression, GO analyses of biological function and cellular component showed that genes within these groups encoded for ion channels (S/ER or plasma membrane) and regulatory proteins that were involved in biological and molecular processes such as Ca2+ signalling and muscle contractility (s-Table 2). Interestingly, genes affected by loss of Brn-3b were also implicated in human diseases since Jensen.disease pathway analysis (Table 1c) showed that the most significant pathways affected by genes upregulated in Brn-3b KO aortas were hypertension (169 genes; NES + 1.48) CAD (128 genes; NES + 1.53).
Consistent with K-means cluster analysis, genes upregulated by loss of Brn-3b were also implicated in pathways that can indirectly affect vascular function including circadian rhythm and entrainment, (68 genes; NES + 1.64); immune function (56 genes; NES + 1.48 that included IL-17 signalling, cytokine receptor interaction, haematopoietic cell lineage), and metabolic diseases (type II diabetes mellitus) (34 genes; NES + 1.45), (Table 1a and Supplementary Table 2a).
On the other hand, genes down-regulated in Brn-3b KO aortas were implicated in controlling VSMC function and related processes (Table 1b), with 68 down-regulated genes (NES −1.64) linked to VSMC contraction; 40 genes (NES-1.75) associated with regulation of actin cytoskeleton and 33 genes (NES −1.79) linked to Apelin signalling pathway. Other down-regulated genes were linked to immune responses [37 reduced genes (NES −1.98) in chemokine signalling and 16 genes (NES −2.09) linked to IL-17 signalling, indicating complex but, as yet unknown functions for Brn-3b in controlling vascular functions.
DEG2 was next undertaken to identify the most significantly up- and down regulated genes, upon loss of Brn-3b. DESeq analyses showed that 59 genes were significantly upregulated and 74 genes down-regulated in Brn-3b KO aorta (Fig. 5a, b). Selected genes shown in Table 2a, b, highlight how genes that were up- or down regulated upon loss of Brn-3b affected key pathways involved in Ca2+ signalling and muscle contraction and is summarised in Fig. 5c, which shows KEGG graph and proposed effects on calcium signalling pathways.

a Heat map showing differential expression of genes (DEG) in Brn-3b KO aortas, compared with WT controls- down-regulated = top panel; up-regulated = bottom panel. b VENN diagram showing the numbers of up and down-regulated genes, using FDR cut-off = 0.1; fold change =1.5 c Schematic diagram showing how genes that are up-regulated or down-regulated in Brn-3b KO aortas are linked in the calcium signalling pathway. Modified KEGG Image was rendered by Pathview with red boxes indicating genes that were increased in Brn-3b KO aortas while green boxes indicate decreased genes and grey indicate unchanged expression.
Notable changes in Ca2+ signalling gene included up-regulation of CaV channels Cacn2d2, Cacna1d & Cacna1h but also RyR2 and Pln [62, 63] with reduced Atp2a1, S100e9 (Calgranulin A), and RyR1. Similarly, changes were detected in genes encoding sarcomeric proteins that affected muscle structure and VSMC contractility. These changes included up-regulation of Mmp12 (Table 2a; Fig. 6a) but also calcium-dependent phospholipase A2, (Pla2g2e) and atrial natriuretic peptide A (Nppa) while there was significant reduction of Eln (Table 2b; Fig. 6a) and Actn3; Mylpf; Myl3. Other upregulated genes were linked to control of circadian processes or metabolic function while many down regulated genes including Cxcr2, IL1 beta, Ccl12, Csf3r and Igsf9 are implicated in the control of immune responses that may indirectly affect vascular function [45]. These data indicate that changes in Brn-3b expression in the vasculature can contribute to contractile dysfunction and CVD.
Subsequent validation studies confirming that genes involved in of Ca2+ signalling and S/ER pathways were significantly and reproducibly de-regulated in independent Brn-3b KO aortas and primary Brn-3b KO VSMC cultures, were particularly important for identifying the underlying mechanisms that drive contractile abnormalities in Brn-3b KO mutants [83, 84].
For instance, raised intracellular Ca2+ in VSMC, which can occur either by Ca2+ influx from extracellular space following activation of activation of Ca2+ channels e.g. L-type voltage-gated Ca2+ (CaV) in the plasma membrane or by Ca2+ release from intracellular store e.g. S/ER via inositol triphosphate (IP3) receptors or via ryanodine receptors (RyR), which contributes to localised Ca2+increases (Ca2+ transient or sparks) [83, 85]. Increased intracellular Ca2+ can then triggers VSMC contraction by driving calmodulin mediated activation of myosin light chain kinase and phosphorylation of myosin light chain (MLC), resulting in actin–myosin interaction [10, 86].
Conversely, membrane re-polarisation and VSMC relaxation depends on reduction of intracellular Ca2+ levels by its re-uptake into intracellular stores by S/ER Ca2+ATPase (SERCA) pumps (encoded by ATP2a genes) [68], or by efflux via plasma membrane Ca2+ (PMCA) pumps, encoded by ATP2b genes [13, 15, 69]. Ca2+ reuptake into the S/ER is also affected by regulatory proteins such as PLN which inhibits SERCA pump activity. Therefore, changes in genes regulating intracellular Ca2+ levels can cause contractile dysfunction in VSMCs [13, 15, 69].
In this regard, increased expression of genes encoding long-acting L‐type CaV channels e.g. Cacna1d and Cacnb2, in Brn-3b KO aortas will raise intracellular Ca2+ and trigger contraction in VSMCs by facilitating Ca2+ influx via plasma membrane. Therefore, such Ca2+ channels are often targeted by Ca2+ channel blockers used to treat patients with hypertension [63, 76, 84]. Similarly S/ER Ca2+ channels are also affected. For instance, while RyR1 is reduced, increased expression of the RyR2 channels in Brn-3b KO aortas will also promote release of Ca2+ from the S/ER into the cytoplasm to increase intracellular Ca2+ while inducing localised transients that also contribute to vascular contraction [13, 83, 85]. As such, increases in these Ca2+ channel genes in Brn-3b KO VSMCs can promote abnormal vascular contractility both at baseline and in response to stimulus, as observed in myography [62, 87]. Moreover, sustained increases in intracellular Ca2+ may also contribute to phenotypic switching from contractile into proliferative VSMCs because raised intracellular Ca2+ can activate NFAT TF (via calcineurin pathway) to drive transcription of genes associated with cell proliferation [74].
On the other hand, loss of Brn-3b may also affect VSMC re-polarisation and relaxation in the aortas because although Atp2a1 was reduced and Atp2a2 levels remain unchanged, increased expression of the potent SERCA pump inhibitor, Pln, will prevent Ca2+ reuptake into the S/ER. Inhibition of SERCA activity combined with enhanced Ca2+ release (via increased Ryr2) will lead to S/ER Ca2+ depletion and contribute to ER stress and UPR [19]. However, reduction in chaperone proteins such as the known Brn-3b target gene Hsp27 (HspB1) [29] as well as other chaperone genes including HspA8, HspH1 and DnaJa1, will prevent adaptive UPR and thereby trigger phenotypic switching from contractile into proliferative VSMCs [20, 69]. This was indeed confirmed in studies using primary VSMC cultures from Brn-3b KO aortas, which displayed significant hyperresponsiveness to ATP stimulation, with increases peak intensity of Ca2+ transients during early stages reflecting increased Ca2+ release from S/ER stores via IP3R and RyR [69].
Such abnormal Ca2+ responses combined with increased proliferation rates in Brn-3b KO VSMC suggests that deregulated intracellular Ca2+ may help to contribute to phenotypic switching of contractile VSMCs into proliferative, synthetic VSMCs [88]. Whilst additional studies will be needed to confirm whether phenotypic switching occurs upon loss of Brn-3b, it is known that increased intracellular Ca2+ can lead to activation of the NFAT TF (via calcineurin pathway), which in turn, activates transcription of genes associated with cell proliferation. Therefore, deregulation of genes involved in Ca2+ signalling, which are caused by loss of Brn-3b, may also contribute to phenotypic and functional changes in the aortic VSMCs and supports important roles for Brn-3b in controlling vascular integrity and function.
These results suggest that Brn-3b is important for regulating genes that control Ca2+ signalling and VSMC contraction in arterial blood vessels and loss of Brn-3b will contribute to contractile dysfunction, which precedes the development of vascular dysfunction including hypertension and subsequent progression to CAD. Therefore, elucidating the effects of loss of Brn-3b and its target genes could provide insight into the molecular basis of early vascular changes and subsequent development vascular dysfunction /damage [89].
Since this study was undertaken using the constitutive Brn-3b KO mutants, it is noteworthy that loss of Brn-3b caused deregulation of genes that are implicated in regulating immune responses, circadian entrainment and metabolic processes, which can indirectly affect vascular function. Although future studies using tissue-specific mutants can help to determine direct and indirect effects of loss of Brn-3b in the vasculature, changes in different subsets of genes are nonetheless interesting and relevant in view of previous studies showing Brn-3b expression and effects of its loss on relevant tissues. For instance, previous studies showing metabolic dysfunction in Brn-3b KO mutants (hyperglycaemia, insulin resistance and increased visceral WAT deposits [26, 30, 35]), means that deregulation of metabolic genes in Brn-3b KO aortas was unsurprising. However, despite careful attempts to remove all excess tissues around the aortas used for RNA sequencing, at this stage it is unclear if such genes are affected by metabolic changes in the mutant aortas or are link to residual PVAT. Nevertheless, these observations are also interesting because adipose tissues can exert profound endocrine effects on BV wall by producing vasoactive modulators such as adipokines and cytokines [90,91,92]. Indeed, common metabolic dysfunction such as obesity and diabetes are commonly linked to increased risk factors of damage to coronary and systemic circulation and subsequent CVD [12]. Since Brn-3b KO PVAT display significant increases in WAT content which produces pro-inflammatory adipokines and cytokines, this is likely to affect the inflammatory milieu and thereby indirectly alter VSMC phenotype and function [60, 90, 93, 94]. In line with this, TEM images showed infiltration of inflammatory cells into the PVAT surrounding Brn-3b KO aortas not seen in PVAT in WT aortas. Inflammatory changes were also confirmed by RNA sequencing data, which showed significant changes in genes linked to pro-inflammatory pathways e.g. IL-17 signalling and cytokine-cytokine receptor interaction pathways, and are implicated in development of vascular dysfunction and CVDs including hypertension and atherosclerosis [2]. Although the link between loss of Brn-3b and deregulated immune responses are still to be determined, Brn-3b is expressed in immune cells e.g. monocytes [34, 95], T cells and PBMCs [96, 97], so it will be interesting to determine if inflammatory effects in Brn-3b KO tissues cause direct or indirect effects on vascular function and transition to vascular diseases [58]. Furthermore, the implications and effects of upregulation of genes that control circadian processes changes in Brn-3b KO aortas remain unknown but since disruption of the circadian rhythm is commonly associated with ageing and risk of vascular, cardiac and metabolic diseases [58, 98, 99], these mutants model could provide some interesting insight into the mechanisms linking such conditions.
In conclusion, the Brn-3b TF represents a novel and important regulator of genes that control Ca2+ in VSMCs and thereby regulate vascular contractility and function. Since Ca2+ signalling pathways are pivotal for controlling multiple cellular functions in different cell types, regulation of such genes by Brn-3b could have wider implications in understanding how this transcriptional regulator controls gene expression and cell fate in other cellular contexts in which Ca2+ pathways are also critical for function. Loss of Brn-3b also appears to be linked to structural and functional changes in the aortas and was strongly linked to hypertension and coronary artery disease either in Jensen.Disease pathway analysis or GWAS data showing that SNPs in Brn-3b genomic region (chromosome 4q31) were associated with increased risk of hypertension and CHD [40,41,42]. Therefore, these novel findings may provide insight into if/how reduction or loss of Brn-3b may drive early vascular changes during early stages of vascular dysfunction and progression to disease. Finally, deregulation of genes involved in other key processes including immune function, circadian regulation and metabolic processes in Brn-3b KO aortas points to complex but as yet unknown mechanisms by which this regulator may indirectly affect vascular function.
Data availability
RNA sequencing datasets generated and analysed during the current study are submitted as fastq files in National Centre for Biotechnology Information (NCBI) sequence read archive (SRA) database repository (https://www.ncbi.nlm.nih.gov/sra) as PRJNA1034077, after release date 2024-05-01. Gene counts used for iDEP analysis are also available from the corresponding author, on reasonable request.
References
Witteman JC, Kannel WB, Wolf PA, Grobbee DE, Hofman A, D’Agostino RB, et al. Aortic calcified plaques and cardiovascular disease (the Framingham Study). Am J Cardiol. 1990;66:1060–4.
Frostegard J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013;11:117.
Bonarjee VVS. Arterial stiffness: a prognostic marker in coronary heart disease. available methods and clinical application. Front Cardiovasc Med. 2018;5:64.
Kamberi LS, Gorani DR, Hoxha TF, Zahiti BF. Aortic compliance and stiffness among severe longstanding hypertensive and non-hypertensive. Acta Inf Med. 2013;21:12–5.
Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, et al. Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study. Hypertension. 2002;39:10–5.
Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–41.
Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:932–43.
Gasser TC. Aorta. In: Ohayon YPaJ, editor. Biomechanics of living organs. Translational epigenetics: Elsevier Inc; 2017. p. 169–91.
Wilson DP. Vascular smooth muscle structure and function. In: Fitridge R, Thompson M, editors. Mechanisms of vascular disease: a reference book for vascular specialists. Adelaide (AU) 2011.
Iyemere VP, Proudfoot D, Weissberg PL, Shanahan CM. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med. 2006;260:192–210.
Shanahan CM, Weissberg PL. Smooth muscle cell heterogeneity: patterns of gene expression in vascular smooth muscle cells in vitro and in vivo. Arterioscler Thromb Vasc Biol. 1998;18:333–8.
Lacolley P, Regnault V, Segers P, Laurent S. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol Rev. 2017;97:1555–617.
Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol. 2011;3:a004549.
Lipskaia LL, RI., Bobe. and Hajjar R. Calcium cycling in synthetic and contractile phasic or tonic vascular smooth muscle cells. Book chapter Sugi H, editor. Current Basic and Pathological Approaches to the Function of Muscle Cells and Tissues - From Molecules to Humans [Internet]. InTech; 2012. Available from: https://doi.org/10.5772/3003.
Liu Z, Khalil RA. Evolving mechanisms of vascular smooth muscle contraction highlight key targets in vascular disease. Biochem Pharm. 2018;153:91–122.
Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–80.
Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Investig. 2000;106:1139–47.
Kapustin AN, Shanahan CM. Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J Physiol. 2016;594:2905–14.
Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y, et al. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-alpha2 in vivo. Arterioscler Thromb Vasc Biol. 2013;33:595–604.
Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–50.
Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines 4. Cell. 1998;92:307–13.
Chakraborty R, Chatterjee P, Dave JM, Ostriker AC, Greif DM, Rzucidlo EM, et al. Targeting smooth muscle cell phenotypic switching in vascular disease. JVSe Vasc Sci. 2021;1–16.
Farooqui-Kabir SR, Diss JK, Henderson D, Marber MS, Latchman DS, Budhram-Mahadeo V, et al. Cardiac expression of Brn-3a and Brn-3b POU transcription factors and regulation of Hsp27 gene expression. Cell Stress Chaperones. 2008;13:297–312.
Budhram-Mahadeo V, Fujita R, Bitsi S, Sicard P, Heads R. Co-expression of POU4F2/Brn-3b with p53 may be important for controlling expression of pro-apoptotic genes in cardiomyocytes following ischaemic/hypoxic insults. Cell Death Dis. 2014;5:e1503.
Maskell LJ, Qamar K, Babakr AA, Hawkins TA, Heads RJ, Budhram-Mahadeo VS. Essential but partially redundant roles for POU4F1/Brn-3a and POU4F2/Brn-3b transcription factors in the develo** heart. Cell Death Dis. 2017;8:e2861.
Mele L, Maskell LJ, Stuckey DJ, Clark JE, Heads RJ, Budhram-Mahadeo VS. The POU4F2/Brn-3b transcription factor is required for the hypertrophic response to angiotensin II in the heart. Cell Death Dis. 2019;10:621.
Budhram-Mahadeo V, Morris PJ, Lakin ND, Theil T, Ching GY, Lillycrop KA, et al. Activation of the alpha-internexin promoter by the Brn-3a transcription factor is dependent on the N-terminal region of the protein. J Biol Chem. 1995;270:2853–8.
Ounzain S, Bowen S, Patel C, Fujita R, Heads RJ, Budhram-Mahadeo VS. Proliferation-associated POU4F2/Brn-3b transcription factor expression is regulated by oestrogen through ERalpha and growth factors via MAPK pathway. Breast Cancer Res. 2011;13:R5.
Lee SA, Ndisang D, Patel C, Dennis JH, Faulkes DJ, D’Arrigo C, et al. Expression of the Brn-3b transcription factor correlates with expression of HSP-27 in breast cancer biopsies and is required for maximal activation of the HSP-27 promoter. Cancer Res. 2005;65:3072–80.
Budhram-Mahadeo VS, Solomons MR, Mahadeo-Heads EAO. Linking metabolic dysfunction with cardiovascular diseases: Brn-3b/POU4F2 transcription factor in cardiometabolic tissues in health and disease. Cell Death Dis. 2021;12:267.
Dennis JH, Budhram-Mahadeo V, Latchman DS. The Brn-3b POU family transcription factor regulates the cellular growth, proliferation, and anchorage dependence of MCF7 human breast cancer cells. Oncogene. 2001;20:4961–71.
Irshad S, Pedley RB, Anderson J, Latchman DS, Budhram-Mahadeo V. The Brn-3b transcription factor regulates the growth, behavior, and invasiveness of human neuroblastoma cells in vitro and in vivo. J Biol Chem. 2004;279:21617–27.
Budhram-Mahadeo V, Moore A, Morris PJ, Ward T, Weber B, Sassone-Corsi P, et al. The closely related POU family transcription factors Brn-3a and Brn-3b are expressed in distinct cell types in the testis. Int J Biochem Cell Biol. 2001;33:1027–39.
Nurminen V, Seuter S, Carlberg C. Primary vitamin D target genes of human monocytes. Front Physiol. 2019;10:194.
Bitsi S, Ali H, Maskell L, Ounzain S, Mohamed-Ali V, Budhram-Mahadeo VS. Profound hyperglycemia in knockout mutant mice identifies novel function for POU4F2/Brn-3b in regulating metabolic processes. Am J Physiol Endocrinol Metab. 2016;310:E303–12.
Cho JH, Mu X, Wang SW, Klein WH. Retinal ganglion cell death and optic nerve degeneration by genetic ablation in adult mice. Exp Eye Res. 2009;88:542–52.
Mu X, Beremand PD, Zhao S, Pershad R, Sun H, Scarpa A, et al. Discrete gene sets depend on POU domain transcription factor Brn3b/Brn-3.2/POU4f2 for their expression in the mouse embryonic retina. Development. 2004;131:1197–210.
Budhram-Mahadeo VS, Irshad S, Bowen S, Lee SA, Samady L, Tonini GP, et al. Proliferation-associated Brn-3b transcription factor can activate cyclin D1 expression in neuroblastoma and breast cancer cells. Oncogene. 2008;27:145–54.
Samady L, Dennis J, Budhram-Mahadeo V, Latchman DS. Activation of CDK4 gene expression in human breast cancer cells by the Brn-3b POU family transcription factor. Cancer Biol Ther. 2004;3:317–23.
Howson JMM, Zhao W, Barnes DR, Ho WK, Young R, Paul DS, et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. 2017;49:1113–9.
Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30.
Shah S, Henry A, Roselli C, Lin H, Sveinbjornsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11:163.
Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114:1241–57.
Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–7.
Verweij N, Ep**a RN, Hagemeijer Y, van der Harst P. Identification of 15 novel risk loci for coronary artery disease and genetic risk of recurrent events, atrial fibrillation and heart failure. Sci Rep. 2017;7:2761.
Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol. 2011;3:a004317.
Dritsoula A, Papaioannou I, Guerra SG, Fonseca C, Martin J, Herrick AL, et al. Molecular basis for dysregulated activation of NKX2-5 in the vascular remodeling of systemic sclerosis. Arthritis Rheumatol. 2018;70:920–31.
** L, Lipinski A, Conklin DJ. A simple method for normalization of aortic contractility. J Vasc Res. 2018;55:177–86.
Feelisch M, Akaike T, Griffiths K, Ida T, Prysyazhna O, Goodwin JJ, et al. Long-lasting blood pressure lowering effects of nitrite are NO-independent and mediated by hydrogen peroxide, persulfides, and oxidation of protein kinase G1alpha redox signalling. Cardiovasc Res. 2020;116:51–62.
Williams SP, Dorn GW 2nd, Rapoport RM. Prostaglandin I2 mediates contraction and relaxation of vascular smooth muscle. Am J Physiol. 1994;267:H796–803.
Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res. 2003;93:656–63.
Ratz PH, Berg KM, Urban NH, Miner AS. Regulation of smooth muscle calcium sensitivity: KCl as a calcium-sensitizing stimulus. Am J Physiol Cell Physiol. 2005;288:C769–83.
Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinforma. 2018;19:534.
Alonso-Carbajo L, Kecskes M, Jacobs G, Pironet A, Syam N, Talavera K, et al. Muscling in on TRP channels in vascular smooth muscle cells and cardiomyocytes. Cell Calcium. 2017;66:48–61.
Tian CJ, Zhang JH, Liu J, Ma Z, Zhen Z. Ryanodine receptor and immune-related molecules in diabetic cardiomyopathy. ESC Heart Fail. 2021;8:2637–46.
Louch WE, Koivumaki JT, Tavi P. Calcium signalling in develo** cardiomyocytes: implications for model systems and disease. J Physiol. 2015;593:1047–63.
Maury E, Ramsey KM, Bass J. Circadian rhythms and metabolic syndrome: from experimental genetics to human disease. Circ Res. 2010;106:447–62.
Gao P, Gao Pan, Choi M, Chegireddy K, Slivano OJ, Zhao J, et al. Transcriptome analysis of mouse aortae reveals multiple novel pathways regulated by aging. Aging. 2020;12:15603–23.
Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, et al. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 1999;100:1134–46.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7.
Wysocka MB, Pietraszek-Gremplewicz K, Nowak D. The role of apelin in cardiovascular diseases, obesity and cancer. Front Physiol. 2018;9:557.
Oloizia B, Paul RJ. Ca2+ clearance and contractility in vascular smooth muscle: evidence from gene-altered murine models. J Mol Cell Cardiol. 2008;45:347–62.
Ghosh D, Syed AU, Prada MP, Nystoriak MA, Santana LF, Nieves-Cintron M, et al. Calcium channels in vascular smooth muscle. Adv Pharm. 2017;78:49–87.
Stojanovic SD, Fuchs M, Kunz M, **ao K, Just A, Pich A, et al. Inflammatory drivers of cardiovascular disease: molecular characterization of senescent coronary vascular smooth muscle cells. Front Physiol. 2020;11:520.
Cuneo AA, Autieri MV. Expression and function of anti-inflammatory interleukins: the other side of the vascular response to injury. Curr Vasc Pharm. 2009;7:267–76.
Schultz K, Murthy V, Tatro JB, Beasley D. Endogenous interleukin-1 alpha promotes a proliferative and proinflammatory phenotype in human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292:H2927–34.
Seshiah PN, Kereiakes DJ, Vasudevan SS, Lopes N, Su BY, Flavahan NA, et al. Activated monocytes induce smooth muscle cell death: role of macrophage colony-stimulating factor and cell contact. Circulation. 2002;105:174–80.
Wu KD, Bungard D, Lytton J. Regulation of SERCA Ca2+ pump expression by cytoplasmic Ca2+ in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2001;280:C843–51.
House SJ, Potier M, Bisaillon J, Singer HA, Trebak M. The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflug Arch. 2008;456:769–85.
Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801.
Owens EA, Jie L, Reyes BAS, Van Bockstaele EJ, Osei-Owusu P. Elastin insufficiency causes hypertension, structural defects and abnormal remodeling of renal vascular signaling. Kidney Int. 2017;92:1100–18.
Kenny D, Polson JW, Martin RP, Paton JF, Wolf AR. Hypertension and coarctation of the aorta: an inevitable consequence of developmental pathophysiology. Hypertens Res. 2011;34:543–7.
Yasmin, O’Shaughnessy KM. Genetics of arterial structure and function: towards new biomarkers for aortic stiffness? Clin Sci. 2008;114:661–77.
Pang X, Sun NL. Calcineurin-NFAT signaling is involved in phenylephrine-induced vascular smooth muscle cell proliferation. Acta Pharm Sin. 2009;30:537–44.
McKenzie C, MacDonald A, Shaw AM. Mechanisms of U46619-induced contraction of rat pulmonary arteries in the presence and absence of the endothelium. Br J Pharm. 2009;157:581–96.
Amberg GC, Navedo MF. Calcium dynamics in vascular smooth muscle. Microcirculation. 2013;20:281–9.
Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharm Rev. 2016;68:476–532.
House RL, Cassady JP, Eisen EJ, Eling TE, Collins JB, Grissom SF, et al. Functional genomic characterization of delipidation elicited by trans-10, cis-12-conjugated linoleic acid (t10c12-CLA) in a polygenic obese line of mice. Physiol Genom. 2005;21:351–61.
Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol. 2011;3:a004242.
Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, et al. Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation. 2010;121:1818–27.
Kleinz MJ, Davenport AP. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul Pept. 2004;118:119–25.
Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y, et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ Res. 2007;101:e32–42.
Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–8.
Fransen P, Van Hove CE, Leloup AJ, Martinet W, De Meyer GR, Lemmens K, et al. Dissecting out the complex Ca2+-mediated phenylephrine-induced contractions of mouse aortic segments. PLoS One. 2015;10:e0121634.
Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–56.
Wray S, Burdyga T. Sarcoplasmic reticulum function in smooth muscle. Physiol Rev. 2010;90:113–78.
Weissberg PL, Cary NR, Shanahan CM. Gene expression and vascular smooth muscle cell phenotype. Blood Press Suppl. 1995;2:68–73.
Williams B. The aorta and resistant hypertension. J Am Coll Cardiol. 2009;53:452–4.
Younes M, Lechago J, Chakraborty S, Ostrowski M, Bridges M, Meriano F, et al. Relationship between dysplasia, p53 protein accumulation, DNA ploidy, and Glut1 overexpression in Barrett metaplasia. Scand J Gastroenterol. 2000;35:131–7.
Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A 1991;13:277–96.
Montani JP, Carroll JF, Dwyer TM, Antic V, Yang Z, Dulloo AG. Ectopic fat storage in heart, blood vessels and kidneys in the pathogenesis of cardiovascular diseases. Int J Obes Relat Metab Disord. 2004;28:S58–65.
Lehman SJ, Massaro JM, Schlett CL, O’Donnell CJ, Hoffmann U, Fox CS. Peri-aortic fat, cardiovascular disease risk factors, and aortic calcification: the Framingham Heart study. Atherosclerosis. 2010;210:656–61.
Chang L, Garcia-Barrio MT, Chen YE. Perivascular adipose tissue regulates vascular function by targeting vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2020;40:1094–109.
Horimatsu T, Kim HW, Weintraub NL. The role of perivascular adipose tissue in non-atherosclerotic vascular disease. Front Physiol. 2017;8:969.
Neme A, Nurminen V, Seuter S, Carlberg C. The vitamin D-dependent transcriptome of human monocytes. J Steroid Biochem Mol Biol. 2016;164:180–7.
Bhargava AK, Li Z, Weissman SM. Differential expression of four members of the POU family of proteins in activated and phorbol 12-myristate 13-acetate-treated Jurkat T cells 1. Proc Natl Acad Sci USA. 1993;90:10260–4.
Ripley BJ, Rahman MA, Isenberg DA, Latchman DS. Elevated expression of the Brn-3a and Brn-3b transcription factors in systemic lupus erythematosus correlates with antibodies to Brn-3 and overexpression of Hsp90. Arthritis Rheum. 2005;52:1171–9.
Takeda N, Maemura K. Circadian clock and vascular disease. Hypertens Res. 2010;33:645–51.
Desvergne B, Michalik L, Wahli W. Transcriptional regulation of metabolism. Physiol Rev. 2006;86:465–514.
Acknowledgements
We express our sincere gratitude to Drs. Markella Ponticos and Caroline Pellet-Many for reagents, advice and assistance with culturing VSMC from rat and mouse aortas, Dr Elizabeth Slavik-Smith for help with TEM imaging and Prof Mike Duchen for support with the calcium imaging. Aortic velocity measurements were done using ultrasound analysis carried out by Dr. Lauren Maskell and Dr. Daniel Stuckey. Tony Brooks (UCL Genomics) provided invaluable input and help with conducting and analysing during RNA sequencing studies.
Funding
This research was funded by the BHF (PG/16/73/32364 and FS/17/8/32664).
Author information
Authors and Affiliations
Contributions
VY and LM undertook most of the experimental studies and analysis and were involved in providing feedback on the manuscript. OP carried out the myography analysis, provided data for this study and provided feedback on the manuscript. VSB-M is the lead author involved in conception and design of the project as well as obtaining funding for undertaking the research. VB-M also helped to plan and supervised experimental studies, undertook analysis of the RNA sequencing data and prepared the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Sergio Lavandero
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yogendran, V., Mele, L., Prysyazhna, O. et al. Vascular dysfunction caused by loss of Brn-3b/POU4F2 transcription factor in aortic vascular smooth muscle cells is linked to deregulation of calcium signalling pathways. Cell Death Dis 14, 770 (2023). https://doi.org/10.1038/s41419-023-06306-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06306-w
- Springer Nature Limited