
Abstract
The pathogenic mechanisms that underlie the progression of remote degeneration after spinal cord injury (SCI) are not fully understood. In this study, we examined the relationship between endoplasmic reticulum (ER) stress and macroautophagy, hereafter autophagy, and its contribution to the secondary damage and outcomes that are associated with remote degeneration after SCI. Using a rat model of spinal cord hemisection at the cervical level, we measured ER stress and autophagy markers in the axotomized neurons of the red nucleus (RN). In SCI animals, mRNA and protein levels of markers of ER stress, such as GRP78, CHOP, and GADD34, increased 1 day after the injury, peaking on Day 5. Notably, in SCI animals, the increase of ER stress markers correlated with a blockade in autophagic flux, as evidenced by the increase in microtubule-associated protein 2 light chain 3 (LC3-II) and p62/SQSTM1 (p62) and the decline in LAMP1 and LAMP2 levels. After injury, treatment with guanabenz protected neurons from UPR failure and increased lysosomes biogenesis, unblocking autophagic flux. These effects correlated with greater activation of TFEB and improved neuronal survival and functional recovery—effects that persisted after suspension of the treatment. Collectively, our results demonstrate that in remote secondary damage, impairments in autophagic flux are intertwined with ER stress, an association that contributes to the apoptotic cell death and functional damage that are observed after SCI.
Similar content being viewed by others

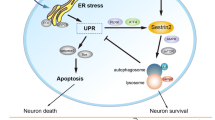

Introduction
Spinal cord injury (SCI) is a devastating condition that affects millions of persons every year worldwide. Consequently, patients suffer from permanent impairments, for which there are no restorative therapies. In SCI, as in other central nervous system (CNS) pathologies, such as stroke, multiple sclerosis, and traumatic brain injury (TBI), the primary insult entails destructive secondary events that can damage cells that were unaffected or marginally affected by the initial insult. Such secondary damage, which occurs from days to years after the insult, is not limited to the lesion site and can involve remote areas that are functionally related to the primary site of injury, leading to remote cell death [1]. Remote degeneration is a multifactorial phenomenon in which many components become active in specific time frames, and despite its clinical importance in determining outcomes in many CNS pathologies, including SCI and TBI [2, Beam-walking test Animals were examined with a fine-motor test paradigm (beam-walking test) at 0 and 1, 3, and 5 days after surgery. In the test, the locomotion of the rats was evaluated pre-operatively and post-operatively using a beam-walking task with an elevated narrow beam (150 cm long × 2.5 cm wide). The worst score (‘0’) was given if the rat was unable to traverse the beam and could neither place the affected limbs on the horizontal surface nor maintain balance. A score of ‘1’ was given if the rat was unable to traverse the beam or to place the affected limbs on the horizontal surface of the beam but was able to maintain balance. A score of ‘2’ was given if the rat was unable to traverse the beam but placed the affected limbs on the horizontal surface of the beam and maintained balance. A score of ‘3’ was given if the rat used the affected limbs in less than half of its steps along the beam. A score of ‘4’ was given if the rat traversed the beam and used the affected limbs to aid with more than 50% of its steps along the beam. A score of ‘5’ was given if the rat traversed the beam and used the affected limbs to aid with less than 50% of its steps along the beam. A score of ‘6’ was given if the rat traversed the beam normally with no more than two-foot slips. The week before surgery, animals were trained to run the narrow beam for a food reward once daily, 5 days per week. All the animals in all groups underwent the motor behavior test to ensure that their performance score before surgery (day 0) was 6. An investigator who was blinded to the experimental groups conducted these experiments. The number of animals used in each experiment is listed in the figure legend section. The numbers of animals used for biochemical analysis (Wb and qRT-PCR), locomotor test, and morphological analysis were based on our previous experience with the techniques and on the basis of a sample size calculation performing a power analysis (G Power 3.1 software). In all cases, we assumed a probability equal to 0.05 and a test power equal to 95%, while Δ and standard deviations were based on previous experiments from our group [8]. For statistical analyses, t-test or one-way, two-way (multiple groups) or repeated-measure analysis of variance (ANOVA) followed, in cases of significance, by a Bonferroni post hoc test was applied. See figure legends for more details. Values of p ≤ 0.05 were considered to be statistically significant. In the box-and-whisker plots, the center line denotes the median value, edges are upper and lower quartiles, whiskers show minimum and maximum values and points are individual experiments. Statistical analyses were carried out by GraphPad Prism 6.0 (GraphPad software for Science, San Diego, CA). All quantitative analyses were conducted blind to the animal’s experimental groups.Experimental design and statistical analysis
Results
SCI induces ER stress and disrupts autophagic flux in remote neurons
To examine the interplay between ER stress and autophagy in remote regions after SCI, we analyzed their kinetics. Because the PERK branch of the UPR is crucial in SCI [25, 39], we considered the mRNA and the protein levels of several key markers of this arm—GRP78, GADD34, and CHOP—in the RN of control (CTRL) and lesioned animals (SCI) at 1, 3, and 5 days after injury (Fig. 1A). The mRNA levels of all ER markers increased in SCI animals, starting from 1 day after the injury, and continued to rise or remained elevated versus CTRL (Fig. 1B). In the protein analysis, we noted a significant increase in GRP78, GADD34, and CHOP in SCI animals compared to CTRL (Fig. 1C).

A Schematic of the protocol used in the study. Adult rats underwent spinal cord injury (SCI), or sham lesion (CTRL). At day 1, 3 and 5 SCI animals were sacrificed and the red nucleus (RN) contralateral to the lesion side was extracted and processed for biochemical analyses or analyzed on fixed-brain sections. B Box-and-whisker plots of GRP78, GADD34 and CHOP mRNA level in control animals (CTRL) and after spinal cord injury (SCI) at various time points after damage (1, 3, and 5 days), expressed as fold over CTRL (N = 5 rats per group; one-way ANOVA, p = 511.1 GRP78; p = 11.87 GADD34; p = 532.3 CHOP) ***p < 0.001, **p < 0.01, *p < 0.05 vs CTRL. C Representative western blot and densitometric box-and-whisker plots from the CTRL and SCI animals at different time points after injury showing the levels (expressed as % of CTRL) of GRP78, GADD34 and CHOP normalized to GAPDH used as loading control (N = 5 rats per group; one-way ANOVA, p < 0.0001 GRP78; p < 0.0001 GADD34; p = 0.0001 CHOP) ***p < 0.001, **p < 0.01, *p < 0.05 vs CTRL. D Representative western blot and densitometric box-and-whisker plots from the different groups showing the levels (expressed as % of CTRL) of LC3, p62, CTSD, LAMP1, and LAMP2 normalized to GAPDH used as loading control (N = 5 rats per group; one-way ANOVA, p < 0.0001 LC3-II; p < 0.0001 p62; p < 0.0001 CTSD; p < 0.0001 LAMP1; p < 0.01 LAMP2). ***p < 0.001, **p < 0.01, *p < 0.05 vs CTRL. In all box-and-whisker plots of the present study the centre line shows the median value, edges are upper and lower quartiles, whiskers show minimum and maximum values, and each point is an individual animal. E Box-and-whisker plots of p62/SQSTM1 mRNA level in CTRL and SCI-sal as fold over CTRL (N = 4 rats per group; Unpaired t-test with Welch's correction p = 0.0125) *p < 0.05.
Further, autophagic activity in remote regions was monitored, based on changes in the autophagosomal protein LC3-II, the autophagy substrate p62/SQSTM1, and the lysosomal markers LAMP1, LAMP2, and cathepsin D (CTSD). Consistent with previous findings on the activation of autophagy after SCI ([8]), we noted a time-dependent increase in the levels of LC3-II (Fig. 1D) that paralleled the upregulation of p62/SQSTM1 at the protein level (Fig. 1D). Considering that in response to various stresses p62 accumulation is also driven by pre-translational regulation, we measured its mRNA level in CTRL and after SCI (SCI 5 days). The results showed that p62 mRNA level increased dramatically in SCI animals compared to CTRL (two fold over CTRL; p < 0.01) (Fig. 1E). These results showed that both p62 mRNA and protein levels increased dramatically in SCI animals compared to CTRL.
Moreover, we examined lysosomal function by measuring CTSD, LAMP1, and LAMP2 levels. As shown in Fig. 1D, CTSD rose significantly in SCI animals compared to CTRL 1 and 3 days after injury and, although slightly decreased at day 5, it remained significantly elevated up to 5 days. Conversely, at the different time points analyzed, LAMP1 rose significantly in SCI animals compared with CTRL 1 and 3 days after injury, while on day 5 it fell down, reaching a level slightly lower than CTRL (Fig. 1D). LAMP2 level remained quite similar to CTRL at 1 day after injury, while on day 3, and more on day 5, it fell down, reaching a level significantly lower than CTRL (Fig. 1D), suggesting that excessive ER stress induced by SCI downregulated lysosomal proteins and, consequently, impairs autophagic flux.
Collectively, our findings show that after SCI, in remote regions, the kinetics of ER stress and autophagy parallel each other, implicating a link between ER stress and the disruption of autophagic flux.
Pharmacological enhancement of UPR restores autophagic flux after SCI
To determine whether the prolonged ER stress observed influences autophagic flux, we treated SCI rats with guanabenz (Fig. 2A), an FDA-approved, centrally acting oral antihypertensive drug that enhances the UPR by kee** prolonged eIF2α phosphorylation [33, 40].

A Schematic of the protocol used in the study. Adult rats underwent spinal cord injury (SCI) received guanabenz (8 mg/Kg i.p. once a day) or saline for 5 days. At day 5 animals were sacrificed and the red nucleus (RN) contralateral to the lesion side was extracted and processed for biochemical analyses or analyzed on fixed-brain sections. B Representative western blot and densitometric box-and-whisker plots from the SCI-saline (SCI-sal) and SCI-Guanabenz (SCI-Guana) treated animals showing the levels (expressed as % of CTRL) of p-eIF2α/eIF2α, GADD34 and CHOPp normalized to GAPDH used as loading control (N = 5 rats per group; Unpaired t test with Welch’s correction p = 0.0057 p-eIF2α/eIF2α; p < 0.0001 GADD34; p < 0.0001 CHOP) ***p < 0.001, **p < 0.01. C Representative western blot and densitometric box-and-whisker plots from the SCI-sal and SCI-Guana groups showing the levels (expressed as % of CTRL) of LC3, p62, CTSD, LAMP1, and LAMP2 normalized to GAPDH used as loading control (N = 5 rats per group; Unpaired t test with Welch’s correction p = 0.7270 LC3-II; p = 0.0001 p62; p = 0.0001 CTSD; p = 0.0001 LAMP1; p < 0.001LAMP2). ***p < 0.001, **p < 0.01. D Confocal Z-stack double immunolabelling for LAMP1 (red) and LC3 (green) puncta in RN neurons from SCI-sal and SCI-Guana animals (scale bar = 10 μm). Single red (LAMP1) or green (LC3) puncta indicates single lysosomes or autophagosomes, respectively. Yellow puncta (merge of red and green) indicates lysosomes fused with autophagosomes (autophagolysosomes). E Box and whisker plots showing the densitometric analyses of LAMP1 and LC3 immunostaining in neurons of RN (N = 5 rats for group; 30 cells/animal; Unpaired t test with Welch’s correction p = 0.0052 LAMP1; p = 0.5029 LC3) ***p < 0.001. F Box and whisker plots showing the co-localization between LAMP1 and LC3 immunostaining in the RN neurons from SCI-sal and SCI-Guana groups. LAMP1-LC3 co-localization is expressed as Pearson’s coefficient of correlation (PCC) (N = 5 rats for group; 30 cells/animal; Unpaired t test with Welch’s correction p < 0.0001) ***p < 0.001.
Five days after daily treatment with guanabenz, we first assessed its efficacy on interfering with ER stress-signaling, such as eIF2α phosphorylation, GADD34, and CHOP levels.
As shown in Fig. 2B, after SCI, guanabenz (SCI-Guana) significantly decreased GADD34 and restored eIF2α phosphorylation over CTRL levels (Fig. 2B). Furthermore, CHOP levels fell significantly in the SCI-Guana versus SCI-sal group, demonstrating that guanabenz has broad effects on ER stress signaling (Fig. 2B). Moreover, in our analysis of autophagy, after SCI, guanabenz did not significantly affect the LC3-II or CTSD levels (Fig. 2C). LC3-II and CSTD levels were elevated in the RN after SCI, and they did not rise further on guanabenz treatment (SCI-sal vs SCI-Guana; Fig. 2C). Conversely, guanabenz significantly altered the levels of p62/SQSTM1, LAMP1, and LAMP2. Specifically, in SCI-Guana animals, p62/SQSTM1 was downregulated compared to the SCI-sal group (Fig. 2C), whereas LAMP1 and LAMP2 levels were upregulated (Fig. 2C).
To confirm these findings, we analyzed the colocalization of LC3 and LAMP1 by confocal analysis to assess autophagosome-lysosome fusion. By immunostaining, LAMP1 expression was significantly higher in the SCI-Guana versus SCI-sal group (Fig. 2D–E), whereas LC3 levels were similar between groups (Fig. 2D–E). Notably, the Pearson’s coefficient of correlation (PCC) for colocalization was higher in the SCI-Guana versus SCI-Sal group (PCCSCI-Guana = 0.328 vs PCCSCI-sal = 0.054; Fig. 2F), suggesting that guanabenz increased the number of autophagolysosomes compared to SCI-sal.
These results demonstrate that ER stress and autophagic flux are closely linked and that the modulation of the ER stress response by guanabenz increases lysosome biogenesis and restores the autophagic flux altered after SCI.
Pharmacological enhancement of UPR mitigates remote apoptotic cell death and improves functional recovery after SCI
Based on the ability of guanabenz to enhance the UPR and restore autophagic flux, we determined its modulatory effects on functional recovery and remote neuronal survival. We first examined whether guanabenz alters the functional-behavioral outcomes that are observed after SCI. Saline-treated (SCI-sal) and SCI-guanabenz-treated (SCI-Guana) animals were evaluated for motor performance using the beam walking test at baseline (before damage, survival time 0) and 1, 3, and 5 days after injury. Notably, after SCI, treatment with guanabenz significantly accelerated functional recovery, based on beam walking test scores (Fig. 3A). Starting at 3 days, the SCI-Guana group had better scores than the SCI-sal group, a difference that was more pronounced 5 days after injury (Fig. 3A).

A Time course of functional recovery measured by Beam-walking test showing the score of SCI-sal and SCI-Guana rats (N = 17 mice/group, m/f = 10/7; Two-way repeated measures ANOVA, (time × treatment: time p < 0.0001; treatment p = 0.0002; interaction: time × treatment p < 0.0001) **p < 0.001; ***p < 0.0001. B Box-and-whisker plots showing the percentage of surviving neurons in RN of SCI-sal and SCI-Guana rats measured by stereological analysis (N = 5 rats/group, m/f = 3/2; Unpaired t test with Welch’s correction p = 0.0036) **p < 0.001. C Representative immunoblots and densitometric box-and-whisker plots showing the level of cytochrome-c (cyt-c) released in the cytoplasm, cleaved Caspase-3 (cl. Casp-3), Bcl-2/Bax ratio normalized to GAPDH used as loading control in the SCI-sal and SCI-Guana groups (N = 5 rats/group, m/f = 3/2; Unpaired t test with Welch’s correction p = 0.0049 Bcl-2/bax ratio; p = 0.0019 cyt-c; p = 0.0005 Caspase-3) **p < 0.001; ***p < 0.0001. D The experimental protocol used in this section to investigate the long-lasting effects of guanabenz treatment. Adult rats underwent spinal cord injury (SCI) received guanabenz (8 mg/Kg i.p. once a day) or saline for 5 days. After that, the treatment was suspended and animals were divided into two groups: a group of animals was left to survive another 16 days after the end of treatment and sacrificed at day 21; the second group was left to survive another 55 days after the end of treatment and sacrificed 60 days after injury. E Time course of functional recovery measured by Beam-walking test showing the score of SCI-sal and SCI-Guana rats (N = 6 rats/group, m/f = 4/2; Two-way RM ANOVA (time × treatment: time p < 0.0001; treatment p = 0.0001; interaction: time × treatment p < 0.0001) **p < 0.001; ***p < 0.0001. F Box-and-whisker plots showing the percentage of surviving neurons in RN of SCI-sal and SCI-Guana rats at different time points (5, 21, and 60 days after injury) measured by stereological analysis (N = 5 mice/group, m/f = 3/2; Unpaired t test with Welch’s correction p = 0.0011) **p < 0.001.
Because neurological recovery following brain injury or SCI is highly influenced by neuronal survival in key brain regions, we reasoned that the neurological improvement that was observed in SCI-Guana animals—i.e., higher beam walking test scores—would be accompanied by greater neuronal survival. By using a quantitative stereological analysis of Nissl-stained neurons, we showed that SCI induced a significant neuronal loss in RN of SCI-sal group (Fig. 3B), further confirmed by TUNEL assay (Supplementary Fig. 1). Furthermore, the percentage of surviving neurons was significantly higher in SCI-Guana versus SCI-sal animals, indicating that guanabenz treatment following SCI significantly mitigates the rate of neuronal degeneration induced by SCI (Fig. 3B). This result prompted us to further determine the modulatory effects of guanabenz on the SCI-induced apoptotic pathway that has been showed effective in inducing neuronal death after SCI [7]. Notably, guanabenz-dependent enhancement of UPR was associated with a significant increase in the Bcl-2/Bax ratio (Fig. 3C) and a reduction in cytochrome-c release from damaged mitochondria (Fig. 3C) and in cleaved caspase-3 (Fig. 3C). Collectively, these findings suggest that guanabenz-dependent enhancement of UPR significantly improves functional recovery in injured animals and halted apoptotic remote cell death due to SCI.
Next, we examined whether guanabenz treatment modulates or maintains functional outcomes and neuronal survival at later time points (21 days and 60 days after injury) (Fig. 3D). To this end, after SCI, we treated animals with guanabenz or saline for 5 days and then suspended the treatment for 16 (SCI-21 Guana or SCI-21 sal) and 55 days (SCI-60 Guana or SCI-60 sal) (Fig. 3D), and their motor performance was evaluated using the beam walking test. Interruption of treatment did not induce long-term motor alterations (Fig. 3E). Whereas scores were slightly worse in SCI-sal animals at 21 and 60 days after injury than at 5 days, such scores were nearly comparable in SCI-Guana animals (Fig. 3E). Notably, the SCI-Guana groups had better scores than SCI-sal animals at 21 and 60 days after injury (Fig. 3E).
Similarly, our stereological analysis revealed that at both time points after injury, the percentage of surviving neurons in the SCI-Guana groups was significantly higher than in the SCI-sal groups (Fig. 3F) but similar compared with at 5 days after guanabenz treatment (Fig. 3F). Conversely, in the SCI-sal group, at 21 and 60 days after injury, this percentage was significantly lower than at 5 days (Fig. 3F), suggesting a faster decline in neuronal survival in SCI-sal versus SCI-Guana animals.
These results suggest that the enhancement of UPR and the restoration of autophagic flux at early time points are crucial in improving and maintaining functional recovery and slowing remote neuronal degeneration induced by SCI.
Guanabenz restores autophagic flux by modulating TFEB
Once determining the long-term effects of guanabenz, we wondered whether the ER stress-induced lysosomal dysfunction that is observed 5 days after injury is due to impaired activity of the MiT/TFE family members, including MITF, TFEB, TFE3, and TFEC, which play crucial roles in the regulation of lysosomal function and autophagy [41, 42]. Given prior evidence that identifies both TFE3 and TFEB as primary factors in the link between ER stress and autophagy [25, 43], we investigated their levels in the different experimental conditions, namely in CTRL, SCI-sal, and SCI-Guana groups.
As shown in Fig. 4A, SCI markedly decreased total levels of TFEB but did not affect the TFE3 levels. After guanabenz treatment, total levels of TFEB were significantly higher than in SCI-sal but lower, although not significant, versus CTRL (Fig. 4A), while total levels of TFE3 were not affected (Fig. 4A). The TFE3 results were confirmed by immunostaining, which showed that TFE3 subcellular expression in the neurons of RN was quite similar in the different experimental conditions considered (Supplementary Fig. 2A), as well as the percentage of RN neurons with nuclear TFE3 expression (Supplementary Fig. 2B).

A Representative immunoblots and densitometric box-and-whisker plots showing the total TFEB and TFE3 levels in CTRL, SCI-sal and SCI-Guana groups (N = 4 rats/group, m/f = 2/2; one-way ANOVA p < 0.0001 TFEB; one-way ANOVA p = 0.1368); **p < 0.01 vs CTRL; §p < 0.05 vs SCI-sal. B Representative immunoblots and densitometric box-and-whisker plots showing the TFEB cytosolic level (normalized to GAPDH) and nuclear level (normalized to H3) in CTRL, SCI-sal and SCI-Guana groups (N = 4 rats/group, m/f = 2/2; one-way ANOVA p = 0.0001); ***p < 0.001 vs CTRL; *p < 0.05 vs CTRL; §§§p < 0.001 vs SCI-sal. C Representative confocal images of TFEB immunofluorescence showing the subcellular compartimentalization of TFEB immunostaining in RN neurons of CTRL, SCI-sal and in SCI-Guana groups (scale bar = 20 μm; inset = 5 μm). D Box and whisker plots showing the percentage of neurons of RN with nuclear expression of TFEB in CTRL, SCI-sal and SCI-Guana (n = 5 sections/rat; N = 5 rats/group; m/f = 3/2; one-way ANOVA p < 0.0001). The process was made off-line and only neurons identified by a clear nuclear profile were included for analysis. Cells presenting nuclear TFEB expression were expressed as percentage of the total number of RN neurons. ***p < 0.001 vs CTRL; §§p < 0.001 vs SCI-sal. E The experimental protocol used for assessing the role of TFEB in restoring the SCI-induced effects. Adult rats underwent spinal cord injury (SCI) received compound C1 (10 mg/Kg i.p. once a day) or saline for 5 days. At day 5 animals were sacrificed and the red nucleus (RN) contralateral to the lesion side was extracted and processed for biochemical analyses or analyzed on fixed-brain sections. F Time course of functional recovery measured by Beam-walking test showing the score of SCI-sal, SCI-Guana and SCI-CompC1 rats (N = 6 mice/group, m/f = 4/2; Two-way RM ANOVA, (time × treatment: time p < 0.0001; treatment p = 0.0002; Interaction: time × treatment p < 0.0001) ***p < 0.001 SCI-sal vs SCI-Guana; §§p < 0.001 SCI-sal vs SCI-CompC1; §§§p < 0.001 SCI-sal vs SCI-CompC1. G Box-and-whisker plots showing the percentage of surviving neurons in RN of SCI-sal, SCI-Guana and SCI-CompC1 rats measured by stereological analysis (N = 5 rats/group, m/f = 3/2; one-way ANOVA p = 0.0003) ***p < 0.0001, **p < 0.001 vs SCI-sal. H Representative confocal images TFEB immunofluorescence from RN showing the compartimentalization of TFEB immunostaining in neurons of SCI-sal, SCI-Guana and SCI-CompC1 groups (scale bar = 20 μm; inset = 5 μm). I Box and whisker plots showing the percentage of neurons of RN with nuclear expression of TFEB in SCI-sal, SCI-Guana, and SCI-CompC1 (n = 5 sections/rat; N = 5 rats/group; m/f = 3/2; one-way ANOVA p < 0.0001) ***p < 0.001 vs SCI-sal. J Representative western blot and densitometric box-and-whisker plots from the SCI-sal, SCI-Guana and SCI-CompC1 groups showing the levels (expressed as % of CTRL) of LC3, p62, LAMP1, and LAMP2 normalized to GAPDH used as loading control (N = 5 rats per group; m/f = 3/2; one-way ANOVA p = 0.4094 LC3-II; p < 0.0001 p62; p < 0.0001 LAMP1; p < 0.001 LAMP2) ***p < 0.0001 vs SCI-sal. §§p < 0.01 vs SCI-Guana.
Considering the TFEB changes observed, in order to clarify the function of TFEB after SCI, we first checked its subcellular localization by Western blot. TFEB is localized in the cytoplasm under normal conditions. However, in response to certain stimuli, such as starvation or injury, TFEB translocates to the nucleus and activates a transcriptional program [44, 45]. As shown in Fig. 4B, SCI markedly increased cytosolic levels of TFEB, and decreased the nuclear levels compared with CTRL (Fig. 4B). After guanabenz treatment, cytosolic levels decreased and nuclear levels were significantly higher than in SCI-sal but lower versus CTRL (Fig. 4B). These results were confirmed by TFEB immunostaining, which showed that TFEB was widely expressed in the cytosol and nuclei of RN neurons in the CTRL group (Fig. 4C) but mainly in the cytosol of neurons and nearly absent in neuronal nuclei after SCI (SCI-sal; Fig. 4C). After guanabenz treatment (SCI-Guana), TFEB was confined primarily to the nuclear compartment (Fig. 4C). Our quantitative analysis of TFEB subcellular localization confirmed that the percentage of neurons with nuclear TFEB expression was lower in the SCI-sal group than in CTRL (Fig. 4D) but significantly higher in SCI-Guana versus SCI-Sal (Fig. 4D), although lower than in CTRL, implicating TFEB in ER stress-mediated dysfunction of autophagy after SCI and guanabenz treatment was effective in modulating TFEB.
To ascertain that guanabenz treatment was directly acting on TFEB, we performed qRT-PCR analysis for the mRNA levels of some TFEB target genes, such as Lamp1, Lamp2, Hexa and ATP6V1A. We found that the mRNA levels of these TFEB target genes were significantly higher in SCI-Guana group compared to SCI-sal (Supplementary Fig. 1C). Collectively, these data indicate that guanabenz was effective in improving TFEB-mediated lysosomal biogenesis.
To validate TFEB results, we treated SCI animals with compound C1 for 5 days (Fig. 4E) and compared its effects to those of saline and guanabenz with regard to functional recovery, neuronal survival, and autophagic flux. Compound C1 binds specifically to TFEB at its N-terminus and promotes its nuclear translocation without inhibiting mTOR activity. By activating TFEB, compound C1 enhances autophagy and lysosome biogenesis in vitro and in vivo [35].
In our analysis of locomotor function, SCI animals that were treated with compound C1 (SCI-CompC1) performed better starting 3 days after injury compared with the SCI-sal group (Fig. 4F) but similarly to the SCI-Guana group (Fig. 4F). Next, we determined the efficacy of compound C1 in promoting neuronal survival and TFEB nuclear translocation in RN neurons. We found that in SCI-CompC1 animals, the percentage of surviving neurons was higher than in SCI-sal (Fig. 4G) but insignificantly lower versus SCI-Guana (Fig. 4G). In a parallel qualitative and quantitative analysis of TFEB localization, the percentage of neurons with nuclear TFEB was significantly higher in SCI-CompC1 versus SCI-sal (Fig. 4H–I) and similar in SCI-Guana animals (Fig. 4H–I).
Notably, with regard to autophagy markers, LC3-II levels were similar between the SCI-CompC1, SCI-Guana, and SCI-sal groups (Fig. 4J), whereas p62/SQSTM1, which was upregulated in RN after SCI, decreased significantly compared to SCI-Sal (Fig. 4J) but similar to the levels observed in the SCI-Guana group (Fig. 4J). Moreover, both LAMP1 and LAMP2 levels rose significantly in SCI-CompC1 compared to SCI-Sal group (Fig. 4J) and, although LAMP1 was insignificantly higher than in SCI-Guana animals (Fig. 4J), LAMP2 levels were not significantly different (Fig. 4J).
Overall, these results suggest that TFEB is crucial in the regulation of autophagic flux in alleviating apoptotic cell death and promoting functional recovery after SCI.
Discussion
The aim of this study was to examine the relationship between ER stress and autophagy and determine whether this crosstalk affects the course of secondary damage in remote regions and outcomes after SCI. Although several studies have evaluated the relationship between ER stress and autophagy in various experimental models of CNS injury [24, 25, 46, 47], their relationship in remote cell death after CNS injury has not been reported.
Using a hemisection model of SCI—a sensitive and reliable paradigm for evaluating supra-spinal changes after spinal injury—we found that ER stress and impaired autophagic flux are intertwined, contributing to remote apoptotic neuronal death. Further, we demonstrated that SCI-induced ER stress directly affects TFEB activity, which is responsible for the lysosomal impairment and blockade in autophagic flux. Enhancement of UPR by guanabenz also restores TFEB activity and, consequently, the autophagic machinery that protects remote neurons from death.
Following SCI, supra-spinal neurons experience a chronic injury state, and the relationship between atrophy and cell death has been largely debated over time generating contradictory results [6]. Counting method, tools to assess death, species, experimental model of SCI employed and age might account for these discrepancies. The cell loss that we observed in rubro-spinal neurons is based on unbiased stereology methods and by molecular analysis of key elements of the apoptotic cascade: cytocrome c release, caspase-3 activation. All of these indices confirmed that SCI-induced activation of the apoptotic cascade, as evidenced by caspase-3 activation and TUNEL assay, clearly indicating the commitment of rubro-spinal neurons irreversibly toward death that was significantly halted by guanabenz treatment.
Cell death due to the accumulation of misfolded proteins in the ER is observed in many pathological conditions, including CNS injury [24, 25, 48,49,50]. To cope with ER stress, damaged cells initiate the UPR, an adaptive signal transduction pathway that, depending on the level of damage, orchestrates the signals that are crucial for the survival or death of cells. Although ER stress is a common hallmark in secondary damage after SCI, whit a massive upregulation of key ER stress markers also rostral to the primary site of injury [51], our study is the first evidence that ER stress occurs even in regions that are remote but functionally connected to the primary injury site.
It is noteworthy that axonal damage activates several signaling pathways that transmit specific molecular messages from the site of injury to the soma of damaged neurons [52]. These mechanisms are essential in sensing the injury locally and for signaling such damage to the cell body to initiate the appropriate somatic responses. Although the mechanisms that induce ER stress in the soma of axotomized neurons are unknown, we can speculate that it can be activated indirectly or directly. In the first case, a warning fast retrograde transmissible signal can reach the soma, where it initiates the protein-folding stress response; alternatively, the ER stress might be induced locally in the axon and subsequently translocated to the cell body. However, we cannot exclude a combination of these two mechanisms, but the extension of this phenomenon and the mechanisms that contribute to the spread of signals near as well as far from the injury site need further investigations.
Autophagy is a highly conserved self-degradation pathway that is involved in the turnover of cytosolic constituents, long-lived proteins, and damaged organelles that are delivered to lysosomes for degradation [53]. Defective autophagy contributes to many diseases, including cancer, cardiovascular diseases, immune-mediated disorders, neurodegenerative diseases [54, 55], and TBI and SCI [56,57,58,59,60,61]. SCI elicits the robust accumulation of autophagosomes, as evidenced by increases in LC3 and p62 at the site of the injury [24, 25] and in distal regions [8].
In our study, we confirmed the accumulation of autophagosomes in RN neurons starting 1 day after injury and peaking at Day 5. Moreover, the accumulation of the substrate protein p62/SQSTM1, paralleling a decrease in the lysosomal proteins LAMP1 and LAMP2 confirmed that the accumulation of autophagosomes resulted from the inhibition of autophagic flux due to impaired lysosome biogenesis. Further, we demonstrated that the disruption in autophagy flux due to altered lysosome biogenesis paralleled the upregulation of ER stress markers. This observation extends previous findings on secondary damage [24, 25], suggesting a causal relationship between ER stress and disruptions in autophagic flux due to impaired lysosome biogenesis as demonstrated on various experimental disease models [27, 62, 40], with neuroprotective effects in various CNS pathologies [33, 64, 65]. Notably, in our model, guanabenz treatment enhanced the UPR and improved autophagic flux. Although we hypothesize that it acts on autophagic flux indirectly, by enhancing UPR, we cannot exclude the possibility that guanabenz has efficacy against various signaling molecules and cells [66, 67], and, thus, also directly on autophagy.
Under stressful conditions, the MiT/TFE family, including MITF, TFEB, TFE3, and TFEC, regulates lysosome function and autophagy [41, 44, 45], the cellular responses to ER stress, and cell fate [68]. Notably, TFEB and TFE3 were recently identified as primary factors in the link between ER stress/UPR and autophagy [25, 69]. Our results extend previous findings and reveal that SCI-induced ER stress alters TFEB activity, but not TFE3, in remote neurons, which was restored by guanabenz through unknown mechanisms. TFEB activity is largely controlled by its subcellular localization, which is regulated primarily by phosphorylation [44, 70]. Our data show that after SCI, TFEB exists largely in the cytoplasm of axotomized neurons of the RN, suggesting that its activity is inhibited. This subcellular localization coincides with the upregulation of ER stress markers, the accumulation of autophagosomes due to lysosomal dysfunction, apoptotic remote cell death, and worse neurological recovery.
After SCI, chronic treatment with compound C1, a curcumin analog and a potent activator of TFEB through direct binding, promoted the nuclear translocation of TFEB and the degradation of the autophagy substrates p62/SQSTM1 and enhanced autophagy and lysosome biogenesis in axotomized neurons of the RN, as shown in other in vivo and in vitro models [35, 71]. Further, in our model, it improved neuronal survival and functional recovery, confirming the function of TFEB as a primary factor in linking the two mechanisms and rendering compound C1 a good neuroprotective drug candidate.
Establishing the link between the sparing of specific neuronal death in a given population and improvements in functional recovery after CNS lesions is challenging. Recovery after SCI requires rearrangements at many levels—in the spinal and supra-spinal regions—and it is not limited to a single brain structure or intracellular signaling pathways. After incomplete SCI, animals spontaneously recover locomotor function, and in our model, untreated SCI rats recovered progressively. In our study guanabenz treatment, as well as compound C1, in parallel with the delay in neurodegeneration, improves spontaneous recovery, based on our beam-walking data. However, we cannot exclude that the effects of guanabenz, and compound C1, might influence injury outcomes by acting on neural centers and cell populations that differ from those that we have considered. However, more work needs to be carried out in order to clarify this aspect.
In conclusion, our findings provide further evidence of the complexity of the mechanism of remote cell death and of the existence of functional interactions between ER stress/UPR, autophagy, and apoptotic remote cell death. Moreover, these findings implicate the recovery of ER proteostasis as a new target for future therapeutic interventions to act on multiple levels to ensure remote neuroprotection after SCI. Additional studies that target the links between ER stress and autophagy are needed to determine each of their contributions—particularly with regard to their kinetics—to the overall changes that are observed after injury to develop treatments for SCI. Furthermore, as it is noteworthy that the autophagic-lysosomal systems, both autophagy and chaperone-mediated-autophagy (CMA), and the UPR are functionally integrated for degrading damaged proteins to maintain cellular homeostasis [72, 73], we cannot exclude the involvement of CMA in our model. Indeed, considering that in several models of diseases [74, 75] CMA activity is upregulated as a compensatory response to autophagy failure, we cannot exclude that at later times after SCI the impaired autophagy can be counterbalanced by CMA. Further studies are needed to elucidate the possible interactions between autophagy, CMA and UPR and their respective roles in the mechanism of remote degeneration.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the corresponding author.
References
Viscomi MT, Molinari M. Remote neurodegeneration: multiple actors for one play. Mol Neurobiol. 2014;50:368–89. https://doi.org/10.1007/s12035-013-8629-x.
Carter AR, Patel KR, Astafiev SV, Snyder AZ, Rengachary J, Strube MJ, et al. Upstream dysfunction of somatomotor functional connectivity after corticospinal damage in stroke. Neurorehabil Neural Repair. 2012;26:7–19. https://doi.org/10.1177/1545968311411054.
Zhang J, Zhang Y, **ng S, Liang Z, Zeng J. Secondary neurodegeneration in remote regions after focal cerebral infarction: a new target for stroke management? Stroke. 2012;43:1700–5. https://doi.org/10.1161/STROKEAHA.111.632448.
Kwon BK, Liu J, Messerer C, Kobayashi NR, McGraw J, Oschipok L, et al. Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc Natl Acad Sci USA. 2002;99:3246–51. https://doi.org/10.1073/pnas.052308899.
Liu P-H, Wang Y-J, Tseng G-F. Close axonal injury of rubrospinal neurons induced transient perineuronal astrocytic and microglial reaction that coincided with their massive degeneration. Exp Neurol. 2003;179:111–26. https://doi.org/10.1006/exnr.2002.8057.
Hassannejad Z, Zadegan S, Shakouri-Motlagh A, Mokhatab M, Rezvan M, Sharif-Alhoseini M, et al. The fate of neurons after traumatic spinal cord injury in rats: a systematic review. Iran J Basic Med Sci 2018;21. https://doi.org/10.22038/ijbms.2018.24239.6052.
Latini L, Bisicchia E, Sasso V, Chiurchiù V, Cavallucci V, Molinari M. et al. Cannabinoid CB2 receptor (CB2R) stimulation delays rubrospinal mitochondrial-dependent degeneration and improves functional recovery after spinal cord hemisection by ERK1/2 inactivation. Cell Death Dis. 2014;5:e1404–e1404. https://doi.org/10.1038/cddis.2014.364.
Bisicchia E, Latini L, Cavallucci V, Sasso V, Nicolin V, Molinari M, et al. Autophagy inhibition favors survival of rubrospinal neurons after spinal cord hemisection. Mol Neurobiol. 2017;54:4896–907. https://doi.org/10.1007/s12035-016-0031-z.
Seranova E, Connolly KJ, Zatyka M, Rosenstock TR, Barrett T, Tuxworth RI, et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017;61:733–49. https://doi.org/10.1042/EBC20170055.
Malik BR, Maddison DC, Smith GA, Peters OM. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol Brain. 2019;12:100. https://doi.org/10.1186/s13041-019-0504-x.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. https://doi.org/10.1038/nature06639.
Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. https://doi.org/10.1016/j.molcel.2010.09.023.
Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–75. https://doi.org/10.1016/j.ceb.2011.05.004.
Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep. 2006;7:880–5. https://doi.org/10.1038/sj.embor.7400779.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. https://doi.org/10.1038/nrm3270.
Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–7. https://doi.org/10.1038/ncb1991.
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. https://doi.org/10.1128/MCB.01453-06.
Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. https://doi.org/10.1371/journal.pbio.0040423.
Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–304. https://doi.org/10.1074/jbc.M607007200.
Yan F, Li J, Chen J, Hu Q, Gu C, Lin W, et al. Endoplasmic reticulum stress is associated with neuroprotection against apoptosis via autophagy activation in a rat model of subarachnoid hemorrhage. Neurosci Lett. 2014;563:160–5. https://doi.org/10.1016/j.neulet.2014.01.058.
Rashid H-O, Yadav RK, Kim H-R, Chae H-J. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–77. https://doi.org/10.1080/15548627.2015.1091141.
Yorimitsu T, Klionsky DJ. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy. 2007;3:160–2. https://doi.org/10.4161/auto.3653.
Yan C, Liu J, Gao J, Sun Y, Zhang L, Song H, et al. Correction: IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis. 2020;11:150. https://doi.org/10.1038/s41419-020-2346-y.
Liu S, Sarkar C, Dinizo M, Faden AI, Koh EY, Lipinski MM, et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015;6:e1582–e1582. https://doi.org/10.1038/cddis.2014.527.
Zhou K, Zheng Z, Li Y, Han W, Zhang J, Mao Y, et al. TFE3, a potential therapeutic target for Spinal Cord Injury via augmenting autophagy flux and alleviating ER stress. Theranostics. 2020;10:9280–302. https://doi.org/10.7150/thno.46566.
González-Rodríguez Á, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179–e1179. https://doi.org/10.1038/cddis.2014.162.
Nakashima A, Cheng S-B, Kusabiraki T, Motomura K, Aoki A, Ushijima A, et al. Endoplasmic reticulum stress disrupts lysosomal homeostasis and induces blockade of autophagic flux in human trophoblasts. Sci Rep. 2019;9:11466. https://doi.org/10.1038/s41598-019-47607-5.
Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. CMM. 2008;8:157–72. https://doi.org/10.2174/156652408784221324.
Cai Y, Arikkath J, Yang L, Guo M-L, Periyasamy P, Buch S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy. 2016;12:225–44. https://doi.org/10.1080/15548627.2015.1121360.
Thangaraj A, Sil S, Tripathi A, Chivero ET, Periyasamy P, Buch S. Targeting endoplasmic reticulum stress and autophagy as therapeutic approaches for neurological diseases. Int Rev Cell Mol Biol. 2020;350:285–325. https://doi.org/10.1016/bs.ircmb.2019.11.001.
Nakka VP, Prakash-babu P, Vemuganti R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: potential therapeutic targets for acute CNS injuries. Mol Neurobiol. 2016;53:532–44. https://doi.org/10.1007/s12035-014-9029-6.
Yin Y, Sun G, Li E, Kiselyov K, Sun D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res Rev. 2017;34:3–14. https://doi.org/10.1016/j.arr.2016.08.008.
Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis. 2014;71:317–24. https://doi.org/10.1016/j.nbd.2014.08.010.
Jiang H-Q, Ren M, Jiang H-Z, Wang J, Zhang J, Yin X, et al. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience. 2014;277:132–8. https://doi.org/10.1016/j.neuroscience.2014.03.047.
Song J-X, Sun Y-R, Peluso I, Zeng Y, Yu X, Lu J-H, et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy. 2016;12:1372–89. https://doi.org/10.1080/15548627.2016.1179404.
Viscomi MT, Florenzano F, Conversi D, Bernardi G, Molinari M. Axotomy dependent purinergic and nitrergic co-expression. Neuroscience. 2004;123:393–404. https://doi.org/10.1016/j.neuroscience.2003.09.030.
Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–83. https://doi.org/10.1080/15548627.2016.1239003.
La Barbera L, Vedele F, Nobili A, Krashia P, Spoleti E, Latagliata EC, et al. Nilotinib restores memory function by preventing dopaminergic neuron degeneration in a mouse model of Alzheimer’s Disease. Prog Neurobiol. 2021;202:102031. https://doi.org/10.1016/j.pneurobio.2021.102031.
Li H, Zhang X, Qi X, Zhu X, Cheng L. Icariin inhibits endoplasmic reticulum stress-induced neuronal apoptosis after spinal cord injury through modulating the PI3K/AKT signaling pathway. Int J Biol Sci. 2019;15:277–86. https://doi.org/10.7150/ijbs.30348.
Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332:91–4. https://doi.org/10.1126/science.1201396.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7. https://doi.org/10.1126/science.1174447.
Yang M, Liu E, Tang L, Lei Y, Sun X, Hu J, et al. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal. 2018;16:31 https://doi.org/10.1186/s12964-018-0242-1.
Bai L, Mei X, Wang Y, Yuan Y, Bi Y, Li G, et al. The role of netrin-1 in Improving functional recovery through autophagy stimulation following spinal cord injury in rats. Front Cell Neurosci. 2017;11:350. https://doi.org/10.3389/fncel.2017.00350.
Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. https://doi.org/10.1126/science.1204592.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB: self-regulation of the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. https://doi.org/10.1038/emboj.2012.32.
Gao B, Zhang X, Han R, Zhang T, Chen C, Qin Z, et al. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta Pharm Sin. 2013;34:657–66. https://doi.org/10.1038/aps.2013.34.
Wang D-Y, Hong M-Y, Pei J, Gao Y-H, Zheng Y, Xu X. ER stress mediated‑autophagy contributes to neurological dysfunction in traumatic brain injury via the ATF6 UPR signaling pathway. Mol Med Rep. 2021;23:247. https://doi.org/10.3892/mmr.2021.11886.
Aufenberg C, Wenkel S, Mautes A, Paschen W. Short communication: spinal cord trauma activates processing of xbp1 mRNA indicative of endoplasmic reticulum dysfunction. J Neurotrauma. 2005;22:1018–24. https://doi.org/10.1089/neu.2005.22.1018.
Ohri SS, Maddie MA, Zhang Y, Shields CB, Hetman M, Whittemore SR. Deletion of the pro-apoptotic endoplasmic reticulum stress response effector CHOP does not result in improved locomotor function after severe contusive spinal cord injury. J Neurotrauma. 2012;29:579–88. https://doi.org/10.1089/neu.2011.1940.
Ohri SS, Hetman M, Whittemore SR. Restoring endoplasmic reticulum homeostasis improves functional recovery after spinal cord injury. Neurobiol Dis. 2013;58:29–37. https://doi.org/10.1016/j.nbd.2013.04.021.
Valenzuela V, Collyer E, Armentano D, Parsons GB, Court FA, Hetz C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 2012;3:e272–e272. https://doi.org/10.1038/cddis.2012.8.
Abe N, Cavalli V. Nerve injury signaling. Curr Opin Neurobiol. 2008;18:276–83. https://doi.org/10.1016/j.conb.2008.06.005.
Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. https://doi.org/10.1101/gad.1599207.
Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. https://doi.org/10.1016/j.cell.2018.09.048.
Zhang J. Autophagy and mitophagy in cellular damage control. Redox Biol. 2013;1:19–23. https://doi.org/10.1016/j.redox.2012.11.008.
Smith CM, Chen Y, Sullivan ML, Kochanek PM, Clark RSB. Autophagy in acute brain injury: feast, famine, or folly? Neurobiol Dis. 2011;43:52–9. https://doi.org/10.1016/j.nbd.2010.09.014.
Lipinski M, Wu J. Modification of autophagy-lysosomal pathway as a neuroprotective treatment for spinal cord injury. Neural Regen Res. 2015;10:892 https://doi.org/10.4103/1673-5374.158344.
Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467–84. https://doi.org/10.1038/nrn.2016.51.
Wolf MS, Bayır H, Kochanek PM, Clark RSB. The role of autophagy in acute brain injury: a state of flux? Neurobiol Dis. 2019;122:9–15. https://doi.org/10.1016/j.nbd.2018.04.018.
Wu J, Lipinski MM. Autophagy in neurotrauma: good, bad, or dysregulated. Cells. 2019;8:693. https://doi.org/10.3390/cells8070693.
Lipinski MM, Wu J, Faden AI, Sarkar C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid Redox Signal. 2015;23:565–77. https://doi.org/10.1089/ars.2015.6306.
Miyagawa K, Oe S, Honma Y, Izumi H, Baba R, Harada M. Lipid-induced endoplasmic reticulum stress impairs selective autophagy at the step of autophagosome-lysosome fusion in hepatocytes. Am J Pathol. 2016;186:1861–73. https://doi.org/10.1016/j.ajpath.2016.03.003.
Fang C, Weng T, Hu S, Yuan Z, **ong H, Huang B, et al. IFN-γ-induced ER stress impairs autophagy and triggers apoptosis in lung cancer cells. OncoImmunology. 2021;10:1962591. https://doi.org/10.1080/2162402X.2021.1962591.
Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, et al. Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma. 2015;32:1608–20. https://doi.org/10.1089/neu.2014.3772.
Sun X, Aimé P, Dai D, Ramalingam N, Crary JF, Burke RE, et al. Guanabenz promotes neuronal survival via enhancement of ATF4 and parkin expression in models of Parkinson disease. Exp Neurol. 2018;303:95–107. https://doi.org/10.1016/j.expneurol.2018.01.015.
Thompson KK, Tsirka SE. Guanabenz modulates microglia and macrophages during demyelination. Sci Rep. 2020;10:19333. https://doi.org/10.1038/s41598-020-76383-w.
Takigawa S, Chen A, Nishimura A, Liu S, Li B-Y, Sudo A, et al. Guanabenz downregulates inflammatory responses via eIF2α dependent and independent signaling. IJMS. 2016;17:674. https://doi.org/10.3390/ijms17050674.
Martina JA, Diab HI, Li H, Puertollano R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci. 2014;71:2483–97. https://doi.org/10.1007/s00018-014-1565-8.
Zhang Z, Qian Q, Li M, Shao F, Ding W-X, Lira VA, et al. The unfolded protein response regulates hepatic autophagy by sXBP1-mediated activation of TFEB. Autophagy. 2021;17:1841–55. https://doi.org/10.1080/15548627.2020.1788889.
Puertollano R, Ferguson SM, Brugarolas J, Ballabio A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018;37. https://doi.org/10.15252/embj.201798804.
Zhuang X-X, Wang S-F, Tan Y, Song J-X, Zhu Z, Wang Z-Y, et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson’s disease models. Cell Death Dis. 2020;11:128. https://doi.org/10.1038/s41419-020-2322-6.
Schreiber A, Peter M. Substrate recognition in selective autophagy and the ubiquitin–proteasome system. Biochimica et Biophysica Acta (BBA) - Mol Cell Res. 2014;1843:163–81. https://doi.org/10.1016/j.bbamcr.2013.03.019.
Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochimica et Biophysica Acta (BBA) - Mol Basis Dis. 2008;1782:691–9. https://doi.org/10.1016/j.bbadis.2008.10.002.
Li W, Zhu J, Dou J, She H, Tao K, Xu H, et al. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat Commun. 2017;8:1763 https://doi.org/10.1038/s41467-017-01609-x.
Li W, Yang Q, Mao Z. Signaling and induction of chaperone-mediated autophagy by the endoplasmic reticulum under stress conditions. Autophagy. 2018:1–3. https://doi.org/10.1080/15548627.2018.1444314.
Acknowledgements
We thank Blue Pencil Science (http://www.bluepencilscience.com/) for editing an English draft of this manuscript. This work was supported by Linea D.1. 2019 and 2020 Università Cattolica del S. Cuore (to MTV) and by the American Alzheimer’s Association (USA) (AARG-18-566270 to MDA), by the Italian Ministry of Health (IT) (Research Grant: RF-2018 -12365527 to MDA and MTV) and by Fondazione Roma (to MDA). LLB was supported by the Strategic University Projects – Young Researcher Independence [YRG2021] by the Università Campus Bio-Medico di Roma (Rome, Italy).
Author information
Authors and Affiliations
Contributions
EB, RM, and AN contributed equally to this work. EB conducted most biochemical experiments and performed animal lesion and treatment, with help from LL, VS. RM, CP, FM, and LLB conducted biochemical experiments on compound C1 animals and reviewed the manuscript. AN conducted the immunostaining and confocal microscopy experiments; RM and AN analyzed data, performed figures, and critically revised drafts of the manuscript; DP, MD, and MTV contributed to supervision, data analysis and provided important intellectual input and contributed to the writing of the manuscript. MTV conceived the idea, devised the study, oversaw the research program, analyzed data, and wrote and revised the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr Flavie Strappazzon
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bisicchia, E., Mastrantonio, R., Nobili, A. et al. Restoration of ER proteostasis attenuates remote apoptotic cell death after spinal cord injury by reducing autophagosome overload. Cell Death Dis 13, 381 (2022). https://doi.org/10.1038/s41419-022-04830-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-04830-9
- Springer Nature Limited
This article is cited by
-
Elamipretide alleviates pyroptosis in traumatically injured spinal cord by inhibiting cPLA2-induced lysosomal membrane permeabilization
Journal of Neuroinflammation (2023)
-
Upregulation of Ca2+-binding proteins contributes to VTA dopamine neuron survival in the early phases of Alzheimer’s disease in Tg2576 mice
Molecular Neurodegeneration (2022)