
Abstract
UVB exposure can contribute to the development of skin cancer by modulating protein tyrosine kinase (PTK) signaling. It has been suggested that UVB radiation increases the ligand-dependent activation of PTKs and induces PTP inactivation. Our recent studies have shown that T-cell protein tyrosine phosphatase (TC-PTP) attenuates skin carcinogenesis induced by chemical regimens, which indicates its critical role in the prevention of skin cancer. In the current work, we report that TC-PTP increases keratinocyte susceptibility to UVB-induced apoptosis via the downregulation of Flk-1/JNK signaling. We showed that loss of TC-PTP led to resistance to UVB-induced apoptosis in vivo epidermis. We established immortalized primary keratinocytes (IPKs) from epidermal-specific TC-PTP-deficient (K14Cre.Ptpn2fl/fl) mice. Immortalized TC-PTP-deficient keratinocytes (TC-PTP/KO IPKs) showed increased cell survival against UVB-induced apoptosis which was concomitant with a UVB-mediated increase in Flk-1 phosphorylation, especially on tyrosine residue 1173. Inhibition of Flk-1 by either its specific inhibitors or siRNA in TC-PTP/KO IPKs reversed this effect and significantly increased cell death after UVB irradiation in comparison with untreated TC-PTP/KO IPKs. Immunoprecipitation analysis using the TC-PTP substrate-trap** mutant TCPTP-D182A indicated that TC-PTP directly interacts with Flk-1 to dephosphorylate it and their interaction was stimulated by UVB. Following UVB-mediated Flk-1 activation, the level of JNK phosphorylation was also significantly increased in TC-PTP/KO IPKs compared to control IPKs. Similar to our results with Flk-1, treatment of TC-PTP/KO IPKs with the JNK inhibitor SP600125 significantly increased apoptosis after UVB irradiation, confirming that the effect of TC-PTP on UVB-mediated apoptosis is regulated by Flk-1/JNK signaling. Western blot analysis showed that both phosphorylated Flk-1 and phosphorylated JNK were significantly increased in the epidermis of TC-PTP-deficient mice compared to control mice following UVB. Our results suggest that TC-PTP plays a protective role against UVB-induced keratinocyte cell damage by promoting apoptosis via negative regulation of Flk-1/JNK survival signaling.
Similar content being viewed by others


Introduction
Skin is the largest organ of the human body. Its function is to provide protection and receive stimuli from the environment; consequently, skin undergoes a great deal of environmental assault. Ultraviolet (UV) irradiation from sunlight is a principal environmental factor that can contribute to the development of all three major types of skin cancer: squamous cell carcinomas, basal cell carcinomas, and cutaneous melanomas. Of the three different ranges of UV, UVB (280–315 nm) radiation is the most significant in causing skin cancers1,2,3. Exposure to UVB radiation damages DNA, giving rise to mutations in any number of genes, including oncogenes and tumor suppressor genes, which can modulate cellular signaling pathways involved in DNA damage repair, growth, apoptosis, and differentiation. Following UVB exposure, irreparable DNA-damaged keratinocytes undergo cell death, or apoptosis, through different pathways that involve the activation of the tumor suppressor gene p53 and cell death receptor signaling. However, aberrant keratinocytes that have managed to survive or bypass apoptosis can start to proliferate during continuous UVB radiation, resulting in the development of UVB-induced skin cancer4,5,6.
T-cell protein tyrosine phosphatase (TC-PTP, encoded by PTPN2) is a tyrosine-specific intracellular non-receptor PTP. Members of the PTP family share a highly conserved motif that is required for dephosphorylation of their target substrate proteins, a reversible posttranslational modification that can active or deactivate proteins. PTPs have been implicated in the pathogenesis of a wide range of human diseases and have become attractive targets for clinical therapies7. TC-PTP is ubiquitously expressed in embryonic and adult tissues8,9. Alternative splicing of PTPN2 generates two distinct forms of TC-PTP: a 45-kDa form known as TC45 (TC-PTPa) and a longer 48-kDa form known as TC48 (TC-PTPb). TC45 is targeted to the nucleus by a bipartite nuclear localization signal in its C terminus10,11. However, our recent studies have shown that TC45 is localized in the cytoplasm of skin keratinocytes and it is translocated to the nucleus in response to UVB irradiation via an AKT/14-3-3σ-dependent mechanism, demonstrating that tissue type is a factor in TC45 subcellular localization12. TC48 is a minor form of TC-PTP that is targeted to the endoplasmic reticulum by its hydrophobic C terminus10,11. TC-PTP modulates various cellular functions, including cell cycle regulation, proliferation, and apoptosis. TC-PTP has been well-studied due to its critical role in the regulation of diabetes and obesity through its ability to modulate insulin and leptin signaling13. For example, neuronal cell-specific TC-PTP-deficient mice showed reduced high-fat-diet-induced weight gain and enhanced leptin sensitivity in the hypothalamus with increased STAT3 phosphorylation after leptin administration, indicating that TC-PTP is involved in the development of leptin resistance via STAT314.
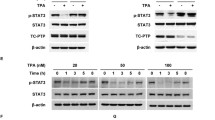
Our work has revealed that TC-PTP is also essential to the skin response to UVB radiation or a two-stage chemical regimen which consists of the carcinogens 7,12-dimethylbenz[a]anthracene (DMBA) and 12-O-tetradecanoylphorbol-13-acetate (TPA). Initial studies of PTPs in skin showed that PTP expression is induced during keratinocyte proliferation and maturation, but expression levels remain unchanged within epidermal tissue15. It has been demonstrated that exposure to acute UV irradiation or treatment with the tumor promoter TPA increases the activation of protein tyrosine kinases, including the epidermal growth factor receptor (EGFR) and the downstream STAT3 signaling pathwayFull size image
To confirm whether TC-PTP directly dephosphorylates Flk-1, the substrate-trap** mutant TC-PTP/D182A, which lacks phosphatase activity, was produced by substituting its essential catalytic residue aspartic acid (D) to alanine (A) and then, a TC-PTP/KO IPK cell line that overexpresses TC-PTP/D182A with a FLAG epitope tag was generated by lentiviral transduction. The mutant TC-PTP/D182A was immunoprecipitated with an anti-FLAG antibody from TC-PTP/D182A-overexpressing TC-PTP/KO IPKs and probed with an anti-Flk-1 antibody. As shown in Fig. 5e, interaction of TC-PTP with Flk-1 was increased following UVB irradiation. Pretreatment with Na3VO4 before UVB exposure blocked their interaction. Immunoprecipitation of Flk-1 in the presence or absence of Na3VO4 confirmed its interaction with TC-PTP after UVB irradiation (Fig. 5f). Consistent with these results, the level of phosphorylated Flk-1 at Y1173 was significantly increased after UVB exposure in the presence of Na3VO4 (Fig. 5g). Together, these results indicate that TC-PTP promotes an apoptotic response in keratinocytes upon UVB irradiation by directly dephosphorylating Y1173 in Flk-1.
JNK is a downstream signal of Flk-1 that is responsible for UVB-induced keratinocyte survival
In keratinocytes, UVB irradiation can initially stimulate ERK and JNK activation to protect cells against UVB-induced apoptosis41,42, which implies that ERK or JNK are potential downstream targets of TC-PTP/Flk-1. Therefore, we examined the levels of phosphorylated ERK and phosphorylated JNK in TC-PTP/WT and TC-PTP/KO IPKs in response to UVB irradiation. Both phosphorylated ERK and phosphorylated JNK expression levels were increased after UVB irradiation (Fig. 6a). While the level of phosphorylated ERK was similarly increased in both types of cells after UVB irradiation, the level of phosphorylated JNK was significantly increased in TC-PTP/KO IPKs compared to TC-PTP/WT IPKs following UVB irradiation (Fig. 6a). To examine whether JNK is involved in UVB-induced apoptosis, IPKs were treated with the JNK-specific inhibitor SP600125 before UVB irradiation. Pretreatment with SP600125 before UVB exposure effectively reduced the level of phosphorylated JNK in both genotypes of IPKs (Fig. 6b). Pretreatment of the JNK inhibitor before UVB exposure reduced cell viability in both TC-PTP/WT and TC-PTP/KO IPKs in comparison to untreated controls. However, the reduction in cell viability of TC-PTP/KO IPKs was higher compared to that in TC-PTP/WT IPKs (Fig. 6c). Consistent with this result, pretreatment of the JNK inhibitor before UVB exposure significantly increased TUNEL-positive apoptotic cells in TC-PTP/KO IPKs to a level comparable with that in TC-PTP/WT IPKs (Fig. 6d, e), which is similar to the results we obtained using Flk-1 inhibitors (Fig. 4g, h). Western blot analysis showed that the levels of both cleaved PARP and cleaved caspase-3 expression were increased in TC-PTP/KO IPKs with pretreatment of the JNK inhibitor, while their expression levels were not changed in TC-PTP/WT IPKs (Fig. 6f). In addition, inhibition of Flk-1 expression or phosphorylation by either siRNA or inhibitors, respectively, reduced the level of phosphorylated JNK in IPKs after UVB irradiation (Fig. 6g, h), demonstrating that JNK is a downstream signaling target of Flk-1.

a Western blot analysis of pJNK and pERK in IPKs from TC-PTP/WT and TC-PTP/KO. Cells were collected at the indicated time after UVB exposure (10 mJ/cm2), lysed, and subjected to SDS-PAGE gel. b Western blot analysis of pJNK in IPKs from both genotypes in response to UVB irradiation. TC-PTP/KO IPKs were incubated with the JNK inhibitor, SP600125 (5 or 20 µM) for 1 h before UVB irradiation (10 mJ/cm2). Cells were incubated with an inhibitor again for 1 h following UVB irradiation at which time the total cell fractions were extracted. c Effect of JNK inhibition on cell viability after UVB exposure. TC-PTP/WT IPKs and TC-PTP/KO IPKs were cultured with SP600125 (5 or 20 µM) for 1 h before UVB irradiation (10 mJ/cm2). Cell viability was analyzed by WST-1 assay 18 h after UVB irradiation. The results are the mean ± standard deviation from three independent experiments. *P < 0.05 by T test for equality of means. d–e TC-PTP/WT IPKs and TC-PTP/KO IPKs were cultured with SP600125 (20 µM) for 1 h before UVB irradiation (10 mJ/cm2). Apoptotic cells were analyzed by TUNEL assay 18 h after UVB irradiation. d Representative TUNEL staining (red) from TC-PTP/WT IPKs and TC-PTP/KO IPKs after UVB exposure. Nuclei were stained with DAPI (blue). Scale bar, 20 µm. e Quantitative analysis of the percentage of TUNEL-positive cells from TC-PTP/WT IPKs and TC-PTP/KO IPKs. The results are the mean ± standard deviation from three independent experiments. *P < 0.05 by T test for equality of means. f Western blot analysis of cleaved PARP and cleaved caspase 3 in TC-PTP/WT IPKs and TC-PTP/KO IPKs. IPKs from both genotypes were cultured with SP600125 (20 µM) for 1 h before UVB irradiation (10 mJ/cm2). Cells were collected at 12 h after UVB irradiation. g–h Western blot analysis for pJNK in IPKs from both genotypes after UVB exposure. g IPKs from both genotypes were cultured and transfected with control or Flk-1 siRNA. After 48 h of siRNA transfection, cells were irradiated with UVB (10 mJ/cm2) and then collected 1 h after UVB irradiation. h IPKs from both genotypes were incubated with SU5416 (2 µM) or ZD6474 (2 µM) for 1 h before UVB irradiation (10 mJ/cm2). Cells were incubated with an inhibitor again for 1 h following UVB irradiation at which time the total cell fractions were extracted.
To examine TC-PTP-mediated regulation of Flk-1/JNK signaling during the response to UVB irradiation in vivo, both TC-PTP/WT and TC-PTP/KO mice were irradiated with UVB, and epidermal cell lysates were prepared for western blot analysis. As shown in Fig. 7a, the levels of phosphorylated Flk-1 and phosphorylated JNK in the epidermis of TC-PTP KO mice were significantly increased in comparison to control mice, especially 1 h after UVB exposure. Collectively, our findings indicate that TC-PTP negatively regulates Flk-1/JNK signaling via direct dephosphorylation of Flk-1 on its Y1173 residue, which contributes to increased epidermal apoptosis in response to UVB exposure (Fig. 7b).

a Western blot analysis of pFlk-1 (Y1173) and pJNK in the epidermis from K14Cre.Ptpn2w/w and K14Cre.Ptpn2fl/fl mice following UVB irradiation at 120 mJ/cm2. Mice were sacrificed at the indicated time after UVB exposure and total epidermal cell lysates were prepared. b Schematic diagram of the regulation of Flk-1/JNK signaling by TC-PTP following UVB irradiation. UVB triggers the phosphorylation of Flk-1 (Y1173) which is a direct target of TC-PTP. In the absence of TC-PTP, Flk-1 phosphorylation on Y1173 can activate JNK signaling, resulting in the promotion of UVB-induced cell survival. TC-PTP contributes to increased epidermal apoptosis in response to UVB exposure by dephosphorylating Flk-1, which may serve as part of an initial protective mechanism against UVB radiation damage.
