
Abstract
The tumour suppressor p53 is a nuclear transcription factor with key roles during DNA damage to enable a variety of cellular responses including cell cycle arrest, apoptosis and DNA repair. JMY is an actin nucleator and DNA damage-responsive protein whose sub-cellular localisation is responsive to stress and during DNA damage JMY undergoes nuclear accumulation. To gain an understanding of the wider role for nuclear JMY in transcriptional regulation, we performed transcriptomics to identify JMY-mediated changes in gene expression during the DNA damage response. We show that JMY is required for effective regulation of key p53 target genes involved in DNA repair, including XPC, XRCC5 (Ku80) and TP53I3 (PIG3). Moreover, JMY depletion or knockout leads to increased DNA damage and nuclear JMY requires its Arp2/3-dependent actin nucleation function to promote the clearance of DNA lesions. In human patient samples a lack of JMY is associated with increased tumour mutation count and in cells results in reduced cell survival and increased sensitivity to DNA damage response kinase inhibition. Collectively, we demonstrate that JMY enables p53-dependent DNA repair under genotoxic stress and suggest a role for actin in JMY nuclear activity during the DNA damage response.
Similar content being viewed by others

Introduction
The human tumour suppressor p53 plays a crucial role in the cellular response to a variety of stressors including DNA damaging and chemotherapeutic agents [1]. As a transcriptional regulator p53 alters gene expression programmes that ultimately impact on cellular outcome that can include cell cycle arrest, apoptosis or DNA repair [2]. Although we still have an incomplete understanding of the mechanisms involved in p53 regulation and how this influences tumour suppression research clearly supports the coordination of DNA repair in being critical for tumour suppression mediated by p53 (reviewed in [3]). Genotoxic stressors (e.g., DNA damaging and chemotherapeutic agents) trigger a myriad of tightly coordinated responses during the DNA damage response (DDR) primarily via ATM, ATR and DNA-PK, members of the phosphatidylinositol 3-kinase-related kinases (PIKK) family [4]. When activated, these kinases phosphorylate multiple targets including p53 [5, 6] and H2AX [7, 8] leading to DNA repair, cell cycle arrest or apoptosis [4]. p53 plays a direct role in DNA repair during the DDR through transcriptional activation of genes involved in a range of DNA repair pathways including for example, TP53I3 (PIG3), XRCC5 (Ku80) and XPC (reviewed in [9]). Defects in p53 function can lead to reduced DNA repair resulting in genomic instability and ultimately tumour development [10]. Interestingly, tumour cells commonly present defects or reduced expression in DNA repair genes which causes the loss of one or more DDR pathways; thus providing the molecular rationale for the use of small-molecule inhibitors of the DDR in cancer therapeutics to exploit these vulnerabilities [10, 11].
The p53 response to DNA damage is influenced by a variety of cofactors that positively and negatively regulate p53 activity [12, 13]. JMY (junction-mediating and regulatory protein) is an actin nucleator and DNA damage-responsive protein. JMY is a member of the WASp (Wiskott–Aldrich Syndrome protein) family of actin nucleation-promoting factors (NPFs) that regulate filamentous (F) actin formation by activating the actin-related protein 2/3 (Arp2/3) complex [14, 15], although JMY uniquely nucleates actin in both an Arp2/3-dependent and independent fashion [15]. JMY localises to both the cytoplasm and the nucleus where cytoplasmic JMY regulates the formation of actin filaments promoting cell motility and invasion [14, 15]. Interestingly, cytoplasmic JMY also increases survival in cells undergoing autophagy, during which JMY’s actin nucleation activity enhances the formation and maturation of the autophagosomes [16]. In response to genotoxic stress, JMY undergoes nuclear accumulation where it has been shown to enhance p53-dependent transcriptional activation of Bax [14, 17]. It is now clear that many of the key players that control actin nucleation in the cytoplasm can also be found in the nucleus and actin plays key roles in nuclear events such as DNA repair [18, 19] and transcriptional regulation [20, 21]. However, we still need to fully understand the impacts of actin nucleators during the DDR and importantly how this might impact on p53 activity in human cancer.
Here, we report that nuclear JMY promotes DNA repair and cell survival during the DDR by enhancing the expression of p53 transcriptional targets involved in DNA repair. JMY-deficient cells exhibit increased DNA damage accumulation and impaired DDR signalling. Importantly, nuclear JMY requires Arp2/3-dependent actin nucleation activity for the efficient clearance of DNA strand breaks. The lack of JMY ultimately increases sensitivity to DNA-damaging agents and, in particular, sensitises cells to DDR inhibition leading to reduced cell survival. Collectively, our results indicate that JMY enhances p53 transcriptional activity to efficiently repair DNA breaks under genotoxic stress and suggest a role of actin nucleation in JMY activity during the DDR.
Results
JMY influences the expression of p53 target genes involved in DNA repair
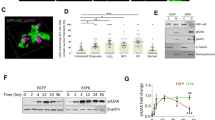
To understand the impact of JMY on gene expression during the DNA damage response we performed transcriptomic analysis (RNA-sequencing) in U2OS osteosarcoma cells under etoposide treatment where JMY undergoes significant nuclear accumulation (Fig. 1ai,ii; [14]). We compared cells treated with either non-targeting or JMY siRNA to identify JMY-mediated impacts on gene expression (Fig. 1b; Fig. S1a, b). Pathway enrichment analyses identified cellular processes that were significantly enriched including the p53 signalling response (q-value = 0.019; Fig. 1c; SI Table 1). JMY depletion resulted in reduced expression of the p53 targets BAX and CDKN1A in support of previous studies [14, 17], but our transcriptomic analysis also identified p53 targets such as Puma/Bbc3 and TIGAR that underwent reduced expression upon JMY depletion (SI Table 2). Closer inspection revealed that while JMY influenced the expression of a range of p53 targets there was enrichment of genes involved in DNA repair with the vast majority of these targets being significantly down-regulated with JMY depletion (Fig. 1d,e; SI Table 1).

a (i) U2OS cells expressing HA-tagged wild-type human JMY (HA-hJMY) were treated with DMSO vehicle (control) or 50 µM etoposide for 6 h. JMY was detected using anti-HA and DAPI was used to visualise nuclei. Scale bar = 10 µm. (ii) Quantification of JMY nuclear versus cytoplasmic accumulation (mean ± SD), N = ≥ 300 cells per treatment, * p < 0.0001, Mann-Whitney test. b Volcano plot represents differentially expressed genes influenced by JMY (q-value < 0.001). Red = upregulated, blue = downregulated and grey = not significant. c Selected enriched KEGG pathways. The threshold was set as Benjamini-Hochberg FDR < 0.05. d Enrichment map representation of p53-related and DNA repair pathways from Reactome database, FDR < 0.05. Nodes and clusters were manually arranged for clarity. e Heatmap showing the relative expression of top p53-downstream targets involved in DNA repair. Changes in gene expression levels are represented as log2(FC). Red = upregulated, blue = downregulated. FDR False Discovery Rate, FC Fold-change expression.
We validated JMY-dependent regulation of p53 targets involved in DNA repair including XRCC5 and XPC by qPCR, confirming that JMY deficiency resulted in a significant reduction in expression during etoposide treatment (Fig. 2a). Conversely, in p53-null Saos2 osteosarcoma cells JMY depletion had little impact on expression (Fig. 2b), confirming that JMY enhances p53-dependent transcription during etoposide-mediated DNA damage. This was reflected in changes to protein expression as siRNA-mediated JMY depletion in U2OS cells resulted in significant reductions in both XPC and XRCC5 (Ku80) protein levels (Fig. 2c, d). The impact of JMY on p53-dependent gene expression was not restricted to a single cell type as we also obtained similar results in p53 wild-type MCF7 breast cancer cells (Fig. S1c). Nor was the effect restricted to etoposide since JMY also undergoes significant nuclear localisation in U2OS cells during treatment with the ultraviolet-radiation mimetic 4NQO (Fig. S2a) and under these conditions JMY depletion also resulted in a reduction in XPC and XRCC5 (Fig. S2b, c).

U2OS (a) and Saos2 (b) cells were transfected with JMY or non-targeting siRNA for 72 h and treated with vehicle (-, DMSO) or etoposide (+, 50 µM) for the last 6 h before harvesting. (i) Changes in gene expression are present as fold mRNA expression relative to non-targeting controls after normalising with GAPDH (mean ± s.e.m.). n = > 3 independent experiments. (ii) Western blot represents JMY knockdown. U2OS cells transfected and treated as in a before protein extraction. (i) Western blots represent XPC (c), XRCC5 (d) levels. (ii) Graph represents expression levels after normalising for actin (mean ± s.e.m.). n = 3–7 independent experiments. e HAP1 parental (WT) and JMY knockout (JMY KO) cells were treated with vehicle (control) or etoposide (500 nM) for 6 h before ChIP. qPCR was performed on ChIP chromatin and results are expressed as fold over IgG (mouse non-specific IgG) after normalising to input levels showing p53 recruitment to XRCC5 (i) or TP53I3 (ii) promoters. n = 2 independent experiments, fold ± SD. ns: not significant, *p < 0.05 and **p < 0.01, Student’s t-test.
Importantly, we compared HAP1 chronic myelogenous leukaemia-derived parental (wild-type p53) and JMY knockout cells (Fig. S3ai), where ablation of JMY resulted in a decrease in both XPC and XRCC5 mRNA and protein (Fig. S3aii, b, c). To determine if JMY influenced p53 recruitment to target genes we performed chromatin-immunoprecipitation (ChIP) in the HAP1 cell lines to allow the comparison in the presence and absence of JMY. In JMY knockout cells there was a marked reduction in p53 recruitment to target genes under etoposide treatment (Fig. 2ei,ii, S3di, ii). Together this demonstrates that nuclear JMY enhances p53 transcriptional activity and recruitment to target genes during the DNA damage response and in particular positively influences the expression of genes involved in DNA repair.
JMY deficiency results in increased DNA damage
The fact that JMY deficiency significantly reduced the expression of key p53 target genes involved in DNA repair suggested that JMY may impact on the accumulation of DNA damage. To measure JMY-mediated impacts on DNA strand breaks we employed alkaline comet assays to directly measure DNA damage in single cells [22]. As expected, both etoposide and 4NQO treatment resulted in a significant accumulation of DNA strand breaks as inferred by comet tail DNA content and length after 16 h (Fig. 3a). While JMY depletion had little impact on the amount of detectable DNA damage under control non-perturbed conditions, under both etoposide and 4NQO treatment JMY depletion resulted in a marked increase in the amount of DNA damage detected (Fig. 3a). These results were recapitulated in both JMY siRNA treated MCF7 (Fig. S4a) and in JMY knockout HAP1 (Fig. 3b) cells. We reasoned that an absence of nuclear JMY during the DDR was negatively impacting on the cells ability to repair DNA and, therefore, overexpressing nuclear JMY should have the converse effect. To test this we used U2OS cells stably overexpressing nuclear localised human JMY (Fig. S4b) to demonstrate that nuclear JMY expression reduced the accumulation of DNA damage during the DDR (Fig. 3c). Moreover, in the absence of p53, JMY depletion did not result in increased DNA damage accumulation (Fig. 3d). In all our results suggest that nuclear JMY is able to enhance p53-dependent DNA repair during genotoxic stress.

a (i) U2OS cells transfected with JMY or non-targeting (NT) siRNA for 72 h before treating with vehicle (control), etoposide (10 µM) or 4NQO (100 nM) for the last 16 h. Comets were stained with Hoechst-33342. (ii) Quantification of the DNA content distributed between the head (black) and tail (grey) of the comet (ii) and the comet tail length (iii), n = 5 independent experiments (mean ± s.e.m.). b HAP1 parental (WT) and JMY knockout (JMY KO) cells were treated with vehicle (control) or etoposide (500 nM) for 16 h. Comet DNA content (ii) and tail length (iii) were calculated as in a, n = 5 independent experiments (mean ± s.e.m.). c i U2OS cells expressing FLAG-NLS-hJMY (NLS-hJMY, NLS) or vector control (vector) were treated as in a. Comet DNA content (ii) and tail length (iii) were calculated as in a, n = 3 independent experiments (mean ± s.e.m.). d Saos2 cells were transfected and treated as in a. Comet DNA content (ii) and tail length (iii) were calculated as in a, n = 3 independent experiments (mean ± s.e.m.). Scale bar = 40 µm. ns: not significant, *p < 0.05, **p < 0.01, Student’s t-test.
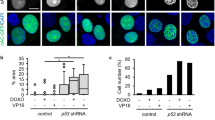
DNA strand breaks induce the formation of DNA damage response foci via the recruitment of repair proteins and these are commonly characterised by markers such as phosphorylated histone H2AX (γH2AX) and 53BP1 [23]. JMY depletion reduced the number of γH2AX and 53BP1 foci detected by immunofluorescence during the DDR (Fig. 4a, b; S5a), as well as decreased total cellular γH2AX levels detected by Western blotting (Fig. S5b). This was also recapitulated in JMY knockout cells where we observed a marked reduction in both γH2AX foci and total levels during the DDR (Fig. 4c; S5c). This suggests that a lack of JMY compromises the cellular response to DNA damage. In support of this, we observed a decrease in total cellular ATM and ATR activity upon JMY depletion or knockout during etoposide treatment (Fig. S5d, e); thus suggesting that JMY enables efficient DDR signalling.

a, b U2OS cells transfected with JMY or non-targeting siRNA for 72 h and treated with vehicle (control) or etoposide (50 µM) for the last 6 h before immunofluorescence. Foci were detected with anti-γH2AX (a) or anti-53BP1 antibodies (b). (ii) Graphs represent the mean number of foci per cell ± s.e.m. for γH2AX (a) or 53BP1 (b), n = 3-4 independent experiments each with N = > 100 cells per condition. (iii) Violin plots represent relative fluorescence intensity for γH2AX (a) or 53BP1 (b) (median and quartiles) N = > 300 cells per condition pooled from n = 3-4 independent experiments. c (i) HAP1 parental (WT) and JMY knockout (JMY KO) cells were treated with either vehicle (control) or etoposide (500 nM) for 6 h before performing immunofluoresence. (ii) Quantification of γH2AX foci per cell (mean ± s.e.m.), n = 5 independent experiments. Scale bars = 10 µm. *p < 0.05 and **p < 0.01, Student’s t-test. #p < 0.0001, Mann-Whitney U test.
JMY-mediated actin nucleation enhances DNA repair
Because nuclear actin is known to play roles in both DNA repair and transcriptional regulation [20] and previous work suggested that actin nucleation may play a role in JMY’s nuclear activities [14], we considered whether JMY’s actin nucleation ability influenced JMY-mediated DNA repair. To address this we used stable cell lines to enable a comparison between nuclear JMY with and without its Arp2/3-dependent and independent actin nucleation activity ([14], Fig. 5a, b, Fig. S6a, b). As observed with NLS-hJMY (Fig. 3c), overexpression of NLS-mJMY resulted in reduced DNA damage during the DDR (Fig. S6a–c). Interestingly, removal of JMY’s entire WH2-domain containing WCA region (NLS-ΔWCA) or ability to mediate Arp2/3-dependent actin nucleation (NLS-W981A) led to increased DNA damage in comparison to wild-type expressing cells (Fig. 5c). This suggested that JMY’s Arp2/3-dependent actin nucleation activity is involved in its ability to influence DNA repair. To explore this further we inhibited Arp2/3 activity (CK666 [24]) in U2OS cells overexpressing NLS-hJMY. Under normal growth conditions Arp2/3 inhibition had no significant impact on DNA damage, while under etoposide treatment, inhibition of Arp2/3 activity prevented the decreased DNA damage seen in the presence of NLS-JMY (Fig. 5d). In addition, JMY’s Arp2/3-dependent actin nucleation activity impacted on the expression of p53 target genes involved DNA repair (Fig. 5e). Thus, JMY-mediated Arp2/3-driven actin nucleation plays a role in DNA repair and this, in part, is mediated through its ability to influence p53-dependent transcription.

a Schematic representation of JMY derivatives lacking the WCA actin nucleation domain (ΔWCA) or presenting a single mutation compromising Arp2/3-dependent actin nucleation (W981A). b Immunofluorescence of U2OS cells stably expressing nuclear JMY and derivatives detected using anti-HA antibody. c U2OS stable cell lines were treated with either vehicle (control) or etoposide (10 µM) for 16 h. Graphs represent mean ± s.e.m. of comet DNA content distributed between the head (black) and tail (grey) (ii) and comets’ tail length (iii), n = 4 independent experiments. d U2OS NLS-hJMY stable cells were treated with either vehicle (control), etoposide (10 µM), and CK666 (100μM) as indicated for 16 h. Comet DNA content (ii) and tail length (iii) were calculated as in c, n = 4 independent experiments (mean ± s.e.m.) e U2OS stable JMY cell lines were treated with etoposide (50 μM) for 6 h before RNA isolation and RT-qPCR. Results represent fold mRNA expression relative to vehicle treatment after normalising with GAPDH (mean ± s.e.m.). n = 4 independent experiments. Scale bars = 40 µm. *p < 0.05, **p < 0.01 and ***p < 0.001 Student’s t-test.
JMY promotes cell proliferation and survival during the DDR
Given that JMY can enhance DNA repair via the p53-response, we explored the impact of JMY on cell fate during the DDR. Short-term JMY depletion had a marked effect on cell proliferation during unperturbed growth conditions with a modest but significant effect under etoposide treatment (Fig. 6a), while JMY knockout cells displayed a more dramatic decrease in proliferation under etoposide treatment (Fig. S7a). This decrease in cell proliferation was reflected in increased cell death during the DDR in both the knockdown and knockout models (Fig. 6b; Fig. S7b, e). Because JMY levels correlate with reduced DDR signalling and increased DNA damage, we reasoned that JMY may influence sensitivity to DDR inhibition. Indeed, under normal growth conditions, JMY deficiency resulted in increased sensitivity to inhibition of ATM, ATR or DNA-PK (Fig. 6c; Fig. S7c), which was further exacerbated during the DDR (Fig. 6d; Fig. S7d).

Cell confluence of U2OS cells transfected with JMY siRNA or non-targeting control and treated with either vehicle (control), etoposide (10 µM) (a) and ATM (ATMi; KU60019 5 µM), ATR (ATRi; AZD6738 5 µM) or DNA-PK (DNA-PKi; M3814 5 µM) inhibitors (c) as indicated. Graphs represent cell confluence as fold change after normalising to time zero images (mean ± SD), n = 3 independent experiments, representative experiment shown. b, d U2OS cells were transfected as in a and treated with vehicle (control), etoposide (as noted) or 4NQO (500 nM) (c), or ATM (ATMi, 10 μM), ATR (ATRi; 10 μM) and DNA-PK (DNA-PKi; 1 μM) inhibitors in the presence or absence of etoposide (10 μM) (d) before collecting for flow cytometry. Graphs represent percentage subG1 (mean ± s.e.m.), n = 3–5 independent experiments. e, f Median mutation count of tumours stratified by low (EXP < −0.5) versus high (EXP > 0.5) JMY mRNA expression levels (i), JMY amplification versus deletion (ii) or stratified by p53 (TP53) mutation status (f), obtained from TCGA pan-cancer dataset. g Kaplan–Meier survival curves for patients whose tumours express low (light grey) or high (dark grey) levels of JMY mRNA, obtained from TCGA pan-cancer study. h During DNA damage, JMY is required for the p53-mediated expression of DNA repair targets, and via its Arp2/3-dependent actin nucleation, impacts on the accumulation of DNA lesions and overall cell survival. *p < 0.05, **p < 0.01 and ***p < 0.001, Student’s t-test.
In patient samples, we found that across all cancer types (TCGA pan cancer), tumours with lower JMY mRNA expression or homozygous deletion contained an increased mutation count (Fig. 6ei, ii). Moreover, further stratification based on p53 (TP53) mutation status revealed that those tumours with higher JMY mRNA levels along with wild-type p53 expression exhibited a significantly lower mutation count (Fig. 6f). Given that JMY reduced DNA damage accumulation, increased resistance to chemotherapeutic drugs, and reduced overall mutation count, we hypothesised that tumours with higher JMY expression might exhibit a more aggressive phenotype resulting in poorer patient survival. Indeed, patients with higher JMY mRNA expression presented substantially lower overall survival (Fig. 6g). Thus, our results suggest that JMY can enhance cell survival during the DNA damage response through impacting on p53-mediated gene expression and DNA repair and this is reflected in patient outcomes in human cancers.
Discussion
The tumour suppressor p53 plays a key role in DNA repair both indirectly by triggering cell cycle arrest as well as directly via transcriptional activation of DNA repair genes [3, 13]. Here we present a novel role for JMY in DNA repair during the DDR and show how the lack of JMY compromises the expression of p53 targets involved in DNA repair and hinders DDR signalling leading to the accumulation of DNA damage. Moreover JMY’s Arp2/3-dependent actin nucleation activity enhances DNA repair and p53 target gene expression. Ultimately depletion of JMY sensitises cells to DDR inhibitors and impacts on cell survival and this is reflected in human cancers where a lack of, or decreased, JMY mRNA expression results in better overall survival and increased tumour mutation count (Fig. 6h).
JMY is an actin nucleator and DNA damage-responsive protein that undergoes nuclear accumulation upon genotoxic stress [14, 17, 25]. JMY supports both Arp2/3-dependent and independent actin nucleation [15] and in the cytoplasm JMY can enhance autophagy to promote cell survival [16]. In the nucleus, JMY influences p53 transcriptional activity where previous studies demonstrated JMY could enhance p53-driven expression of Bax [17]. Our transcriptomic analysis supports a wider role for JMY in transcriptional regulation and ability to influence p53-dependent gene expression, in particular genes involved in DNA repair. A multitude of factors regulate target gene selection by p53, including p53 activation and stability which can be influenced by post-translational modifications, p53 cofactors and binding proteins [2]. p53 is regulated by all three DDR kinases (ATM, ATR and DNA-PK) resulting in stabilisation and enhanced nuclear localisation [2]. JMY’s impact on DDR signalling influences p53 recruitment to target promoters (Fig. 2e). Further, JMY’s impacts on p53-dependent gene expression will also influence downstream DDR signalling as well as DNA repair. XRCC5 (Ku80), for example, together with XRCC6 (Ku70) forms the DNA-binding Ku heterodimeric complex that forms a scaffold to recruit other repair proteins to DNA damage sites, including the DDR kinase DNA-PK [26]. A reduction in DNA-PK recruitment to damaged chromatin in the absence of JMY could explain the sensitivity of JMY knockout cells to DNA-PK inhibition (Fig. S7d). Overall, we have expanded our understanding of the role of JMY during the DDR to show that JMY reduces DNA damage accumulation by both positively impacting on p53-dependent transcriptional activity and DDR signalling.
Although incompletely understood, we know that different stressors can influence the subcellular localisation of JMY. For example, certain genotoxic stressors result in JMY nuclear accumulation (Fig. 1a; Fig. S2a; [25]), while metabolic stressors such as starvation lead to JMY association with cytoplasmic autophagosomes [16]. Recently, JMY was shown to influence p53-dependent apoptosis through cytoplasmic Arp2/3-dependent actin nucleation and influence several steps in the mitochondrial-dependent apoptotic process [27], thus adding further complexity to JMY’s cytoplasmic role as well as its overall impact on cell fate. It is clear that JMY can influence cell survival via a number of different means and it is likely that differences in the type of stressor and duration and dose will impact on JMY’s subcellular localisation and thus activity in the cell. Studies have shown that p53 activity and outcome is influenced by the duration and type of stressor. For example, pulsating p53 levels have been shown to activate a transient expression of DNA repair and cell cycle arrest genes [28, 29], while more sustained expression of p53, leads to the activation of pro-apoptotic genes [29]. Thus it is likely that for a shift between DNA repair to apoptotic p53-dependent gene expression, the levels of p53 must exceed a time-dependent threshold [29, 30]. Further studies are required to refine our understanding of how temporal and dose-dependent effects influence JMY-mediated impacts on p53 activity during the DNA damage response and how this modulates gene expression programmes to influence outcome. Moreover, JMY promotes cell survival through its influence on nuclear activities such as transcriptional regulation as well as through its positive impact on autophagy [16]. It will be relevant for future studies to assess the impact of JMY on autophagy during the DDR as, for example, etoposide can enhance autophagy [41] using the reference transcriptome (v82). DESeq2 (GALAXY Version 1.1.0) [42] was used to normalise and calculate the differential transcript expression between JMY and non-targeting siRNA cells. A final list of differentially expressed genes was obtained using q-value < 0.001.
Pathway enrichment analysis was performed following Reimand and colleagues’ protocol [43]. Briefly, differentially expressed genes were used as input for the g:GO analysis from gProfiler [44] with a significant threshold of Benjamini-Hochberg FDR < 0.05. Enriched pathways and gene ontologies were obtained from the KEGG, REACTOME and the Gene Ontology Consortium databases, respectively.
To explore the role of JMY in patient outcomes in human cancers, we used the cBioPortal for Cancer Genomics database (http://www.cbioportal.org; [45]). Data was obtained using the ICGC/TCGA pan-cancer cohort [46], including 2,922 samples from 2583 patients. Samples were manually grouped based on (i) JMY expression levels (mRNA expression z-scores, high: EXP > 0.5 or low: EXP < −0.5) or JMY copy number (amplification: AMP or homozygous deletion: HOMDEL). Groups were further split based on p53 mutation status (wild-type: WT or mutant: mut) using cBioportal Onco Query Language. Clinical data were retrieved, including Kaplan–Meier patient survival curves and mutational counts.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.0.2. All data were tested for normal distribution. At least three independent experiments were performed, and individual data points are represented. Results with error bars represent mean ± standard error of the mean (s.e.m.), unless otherwise specified in the figure legends. Statistical analyses were performed using at least 3 independent biological repeats (n values). For quantification of fluorescence in Figs. 1a, 4aiii, 4biii, S2a, cell numbers (N values) were used for statistical analyses by pooling the data from at least 3 independent biological repeats with a minimum of 100 cells/images per condition for each repeat. The differences between two groups were analysed by unpaired two-tailed Student’s t-test for normalised data, and Mann–Whitney U test for non-normalised data. All values were considered significant with p-value < 0.05.
Data availability
RNA-sequencing data have been deposited to Array Express under accession number 513 E-MTAB-12059. Constructs generated in this study are available upon request.
References
Vaddavalli PL, Schumacher B. The p53 network: cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022;38:598–612.
Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. 2020;20:471–80.
Thomas AF, Kelly GL, Strasser A. Of the many cellular responses activated by TP53, which ones are critical for tumour suppression? Cell Death Differ. 2022;29:961–71.
Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–17.
Banin S, Moyal L, Shieh SY, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7.
Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9.
Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates Histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7.
Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62.
Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med. 2016;6:1–16.
Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168:644–56.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome Atlas. Cell Rep. 2018;23:239.
Coutts AS, La, Thangue NB. The p53 response: Emerging levels of co-factor complexity. Biochem Biophys Res Commun. 2005;331:778–85.
Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199–210.
Coutts AS, Weston L, La Thangue NB. A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc Natl Acad Sci USA. 2009;106:19872–7.
Zuchero JB, Coutts AS, Quinlan ME, La Thangue NB, Mullins RD. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol. 2009;11:451–9.
Coutts AS, La Thangue NB. Actin nucleation by WH2 domains at the autophagosome. Nat Commun. 2015;6:1–9.
Shikama N, Lee CW, France S, Delavaine L, Lyon J, Krstic-Demonacos M, et al. A novel cofactor for p300 that regulates the p53 response. Mol Cell. 1999;4:365–76.
Caridi CP, Plessner M, Grosse R, Chiolo I. Nuclear actin filaments in DNA repair dynamics. Nat Cell Biol. 2019;21:1068–77.
Schrank BR, Aparicio T, Li Y, Chang W, Chait BT, Gundersen GG, et al. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature. 2018;559:61–66.
Kyheröinen S, Vartiainen MK. Nuclear actin dynamics in gene expression and genome organization. Semin Cell Dev Biol. 2020;102:105–12.
Wei M, Fan X, Ding M, Li R, Shao S, Hou Y, et al. Nuclear actin regulates inducible transcription by enhancing RNA polymerase II clustering. Sci Adv. 2020;6:eaay6515.
Singh NP. The comet assay: Reflections on its development, evolution and applications. Mutat Res Rev Mutat Res. 2016;767:23–30.
Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613–20.
Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, et al. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009;460:1031–4.
Coutts AS, Boulahbel H, Graham A, La Thangue NB. Mdm2 targets the p53 transcription cofactor JMY for degradation. EMBO Rep. 2007;8:84–90.
Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33.
King VL, Leclair NK, Coulter AM, Campellone KG. The actin nucleation factors JMY and WHAMM enable a rapid Arp2/3 complex-mediated intrinsic pathway of apoptosis. PLOS Genet. 2021;17:e1009512.
Hafner A, Stewart-Ornstein J, Purvis JE, Forrester WC, Bulyk ML, Lahav G. P53 pulses lead to distinct patterns of gene expression albeit similar DNA-binding dynamics. Nat Struct Mol Biol. 2017;24:840–7.
Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 dynamics control cell fate. Science. 2012;336:1440–4.
Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell. 2016;165:631–42.
Chen TY, Huang BM, Tang TK, Chao YY, **ao XY, Lee PR, et al. Genotoxic stress-activated DNA-PK-p53 cascade and autophagy cooperatively induce ciliogenesis to maintain the DNA damage response. Cell Death Differ. 2021;28:1865–79.
Sarkar K, Han SS, Wen KK, Ochs HD, Dupre L, Seidman MM, et al. R-loops cause genomic instability in T helper lymphocytes from patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2018;142:219–34.
Wang T, Du XH, Hong Y, Hong X, Fan L, Zhou JW, et al. WASH interacts with Ku to regulate DNA double-stranded break repair. iScience. 2022;25:103676.
Coutts AS, Pires IM, Weston L, Buffa FM, Milani M, Li JL, et al. Hypoxia-driven cell motility reflects the interplay between JMY and HIF-1alpha. Oncogene. 2011;30:4835–42.
Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV. OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2014;2:457–65.
Schindelin, J, Arganda-Carreras, I, Frise, E, Kaynig, V, Longair, M, Pietzsch, T, et al. Fiji: An open-source platform for biological-image analysis. Nature Methods: Nature Publishing Group; pp. 676-82 (2012).
Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 2013;3:71–85.
Herbert AD, Carr AM, Hoffmann E. FindFoci: a focus detection algorithm with automated parameter training that closely matches human assignments, reduces human inconsistencies and increases speed of analysis. PLoS One. 2014;9:e114749.
Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinforma. 2021;22:433.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:1–13.
Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21.
Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc. 2019;14:482–517.
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. G:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47:W191–W198.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2012;2:401–4.
Consortium ITP-CAoWG. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93.
Acknowledgements
The authors would like to thank Glen Kirkham (Nottingham Trent University) and Eric O’Neill (University of Oxford) for valuable reagents. This work was supported by a Wellcome Trust Seed Award (ASC) and Nottingham Trent University QR funding (ASC, EB) and VC-funded PhD scholarship (IRP).
Author information
Authors and Affiliations
Contributions
Project conception, funding acquisition, and supervision (ASC), data generation and analysis (IRP, ASC, EB), and manuscript preparation (ASC, IRP).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodriguez-Pastrana, I., Birli, E. & Coutts, A.S. p53-dependent DNA repair during the DNA damage response requires actin nucleation by JMY. Cell Death Differ 30, 1636–1647 (2023). https://doi.org/10.1038/s41418-023-01170-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-023-01170-9
- Springer Nature Limited