
Abstract
Photoreceptor apoptosis is recognized as one key pathogenesis of retinal degeneration, the counteraction of which represents a promising approach to safeguard visual function. Recently, mesenchymal stem cell transplantation (MSCT) has demonstrated immense potential to treat ocular disorders, in which extracellular vesicles (EVs), particularly exosomes, have emerged as effective ophthalmological therapeutics. However, whether and how MSCT protects photoreceptors against apoptotic injuries remains largely unknown. Here, we discovered that intravitreal MSCT counteracted photoreceptor apoptosis and alleviated retinal morphological and functional degeneration in a mouse model of photoreceptor loss induced by N-methyl-N-nitrosourea (MNU). Interestingly, effects of MSCT were inhibited after blockade of exosomal generation by GW4869 preconditioning. Furthermore, MSC-derived exosomal transplantation (EXOT) effectively suppressed MNU-provoked photoreceptor injury. Notably, therapeutic efficacy of MSCT and EXOT on MNU-induced retinal degeneration was long-lasting as photoreceptor preservance and retinal maintenance were detected even after 1–2 months post to injection for only once. More importantly, using a natural occurring retinal degeneration model caused by a nonsense mutation of Phosphodiesterase 6b gene (Pde6bmut), we confirmed that MSCT and EXOT prevented photoreceptor loss and protected long-term retinal function. In deciphering therapeutic mechanisms regarding potential exosome-mediated communications, we identified that miR-21 critically maintained photoreceptor viability against MNU injury by targeting programmed cell death 4 (Pdcd4) and was transferred from MSC-derived exosomes in vivo for functional regulation. Moreover, miR-21 deficiency aggravated MNU-driven retinal injury and was restrained by EXOT. Further experiments revealed that miR-21 mediated therapeutic effects of EXOT on MNU-induced photoreceptor apoptosis and retinal dysfunction. These findings uncovered the efficacy and mechanism of MSCT-based photoreceptor protection, indicating exosomal miR-21 as a therapeutic for retinal degeneration.
Similar content being viewed by others

Introduction
Photoreceptor apoptosis is recognized as one of the major form of photoreceptor death and one important contributor to visual loss in retinal degenerative disorders [1, 2], such as inherited retinal dystrophies encompassing the retinitis pigmentosa (RP) [3]. Recently, mesenchymal stem cell transplantation (MSCT) has demonstrated immense potential to ameliorate a variety of ocular dysfunctions [4, 5]. Particularly, MSCT retards retinal degeneration with the prevention of loss of retinal ganglion cells (RGCs) and retinal pigment epithelium (RPE) [1, 2]. Extensively efficient to ameliorate retinal traumatic, ischemic, and oxidative injuries [7, 18] as well as retarding inflammatory and diabetic retinopathies [8, 52], MSCT represents promising cell-based therapeutics which also rescues photoreceptor deficiency in mice with genetic defects [9,10,11]. Notably, there are studies reporting retinal damages and ocular complications induced by intravitreal or subretinal MSCT, indicating that stem cell delivery should still be applied with caution [53, 54]. In this study, we for the first time revealed that MSCT protected photoreceptors against specific apoptotic stimulus in vivo, and further identified EXOT as cell-free alternative strategy to overcome potential drawbacks of cell transplantation with the beneficial effects preserved. Moreover, we discovered that a single intravitreal injection of MSCs or exosomes has a long-lasting protection of photoreceptors in both the MNU pharmacological model and the Pde6bmut genetic model with ameliorated loss of either scotopic (mixed rod and cone responses) or photopic (cone-mediated) ERG responses, which therefore provides promise for establishing novel therapeutics of retinal degeneration.
It is recognized that MSCT maintains tissue homeostasis through either paracrine effects to establish beneficial microenvironments or through inhabitation in recipient tissues to replenish deficient cells [14, 55]. After intravitreal transplantation, exogenous MSCs have been traced to remain in the vitreous body, in which no retinal incorporation has been reported [56,57,58]. Nevertheless, it has been claimed in MSCT treating a mouse model of RP that transplanted MSCs morphologically integrated into the RPE, while not being detected in other retinal layers [10]. Additionally, in vitro experiments indicated regulation of MSCs by the retinal microenvironment, in that RPE cell-conditioned medium and photoreceptor outer segments stimulate differentiation of MSCs toward the RPE cell phenotype, but the in vivo functional evidence of MSC replacing retinal cells is lacking [59]. Here, we showed that the biodistribution of injected MSCs was primarily at the ONL, indicating close functional correlationships with recipient photoreceptors and potential needs of damaging photoreceptors for recovery. However, MSCs were able to release exosomes for therapeutic effects at the ONL, and effects but not biodistribution of MSCs were diminished upon blockade of exosomal generation. Therefore, it can be expected that MSCT improves retinal integrity and function via paracrine effects, as also confirmed by the identification of various neuroprotective and anti-inflammatory factors secreted, such as nerve growth factor, basic fibroblast growth factor, and tumor necrosis factor alpha-stimulated gene-6 [8, 16]. It has additionally been reported that MSC-derived EVs enhance functional recovery of retina after ischemia-reperfusion damages and optic nerve crush injuries, and that transplanted EVs can be uptaken by RGCs and microglia [17, 18]. In this study, we further revealed that EXOT efficiently and continuously counteracted phototoxin-induced and Pde6bmut retinal degeneration by targeting photoreceptors, adding another dimensional to the current knowledge of paracrine-based MSC therapy of retinal disorders. Whether the injected MSCs can also proliferate and differentiate toward photoreceptors in vivo remain to be investigated in future studies.
Shuttle of microRNAs by EVs have been widely proved as significant contributors to tissue homeostasis and feasible therapeutics for diseases [23,24,25]. In the ocular system, it has been reported that EV-mediated microRNA communication may influence posttranscriptional regulation of retinal development, and that microRNAs carried by circulating EVs can be used as prognostic biomarkers for retinopathy [20, 21]. microRNA-dependent mechanisms have also been implicated in MSC-derived EVs to treat corneal fibrosis and optic nerve injury [17, 60]. In this study, we first identified an individual microRNA, miR-21, as a contributor to photoreceptor viability in vivo and a mediator of therapeutic effects of EXOT on retinal dysfunction. Notably, miR-21 has been previously documented mainly as a negative regulator of retinal health, such as inhibition of neovascularization in the ischemic retina by targeting tissue inhibitor of metalloproteinase 3 [61], induction of Müller cell gliosis after optic nerve crush by regulating glial fibrillary acidic protein [39], promotion of autoimmune uveoretinitis by targeting interleukin-10 [62], and facilitation of the progression of retinoblastoma by targeting phosphatase and tensin homolog [63]. Besides, previous studies have shown that miR-21 can induce cell apoptosis by targeting S-phase kinase-associated protein 2 and B-cell lymphoma-2 [64, 65]. Our results showing miR-21 protection against retinal disorders only join with another study demonstrating that the miR-21/Pdcd4 axis regulates MSC-induced neuroprotection in glaucoma, indicating target-specific regulatory effects based on Pdcd4 inhibition [48]. Actually, miR-21 inhibition of Pdcd4 expression for antiapoptotic effects has additionally been reported in other systems [66, 67]. The detailed function and mechanisms of Pdcd4 in regulating photoreceptors and retinal degeneration in vivo remains to be elucidated in future studies.
Materials and methods
Animals
Animal experiments were performed following the Guidelines of Intramural Animal Use and Care Committees of Fourth Military Medical University, **’an Jiaotong University and the ARRIVE guidelines. Twelve-week WT C57BL/6 mice and miR-21−/− mice (C57BL/6 background) (weight, 20–22 g; three male or female mice per cage) were purchased from the Jackson Laboratory, as we have used in previous studies [42, 43]. Two-week-old male or female Pde6bmut mice with a nonsense mutation of Pde6b gene on a C57BL/6J background that result in the failure of protein production [38] were obtained from TC at Department of Clinical Medicine, Fourth Military Medical University. Mice were maintained with good ventilation and a 12-h light/dark cycle, and were kept feeding and drinking ad libitum.
Cell culture
Isolation and culture of MSCs from mouse bone marrow were as previously described [68]. Briefly, whole bone marrow cells were seeded, incubated overnight, and rinsed with phosphate buffer saline (PBS) to remove the non-adherent cells. The adherent cells were cultured with alpha-Minimum Essential Medium supplemented with 20% fetal bovine serum (FBS), 2-mM l-glutamine, 100-U/ml penicillin, and 100-g/ml streptomycin (all from Invitrogen, USA) at 37 °C in a humidified atmosphere of 5% CO2. Primary MSCs were digested with 0.25% trypsin (MP Biomedicals, USA) and passaged for the following experiments after seeding at appropriate densities.
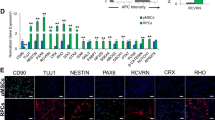
MSCs were verified according to the current standard [32, 33]. For flow cytometric analysis of the surface markers, MSCs at the first passage were collected and suspended in PBS supplemented with 3% FBS at 1 × 106 cells/ml. MSCs were added with FITC-conjugated anti-mouse CD11b antibody (11-0112-82; eBioscience, USA), PE-conjugated anti-mouse CD29 antibody (12-0299-42; eBioscience, USA), FITC-conjugated anti-mouse CD34 antibody (11-0341-82; eBioscience, USA), PE-conjugated anti-mouse CD45 antibody (12-0451-82; eBioscience, USA), PE-conjugated anti-mouse CD31 antibody (12-0311-82; eBioscience, USA), and FITC-conjugated anti-mouse Stem cell antigen 1 (Sca1) antibody (11-5981-82; eBioscience, USA) at concentrations of 1:100. Nonimmune immunoglobulin of the same isotype was used as the NC. MSCs were incubated in 4 °C for 30 min in dark, and then washed twice with PBS supplemented with 3% FBS. The percentages of positively stained cells were determined with a flow cytometer (FACSAria; BD Biosciences, USA) equipped with the FACSDiva Version 6.1.3 software. For cell viability examinations, the seeded MSCs at the first passage were incubated with 20-μl 5-mg/ml methyl thiazolyl tetrazolium (MP Biomedicals, USA) for 4 h [55, 69]. The precipitates were extracted with 180-μl dimethyl sulfoxide (DMSO) and the absorbance was measured at the optical density of 490 nm. Cell viability fold changes were calculated accordingly. For osteogenic differentiation, the seeded MSCs at the first passage were induced in media containing 100-μg/ml ascorbic acid (MP Biomedicals, USA), 2-mM β-glycerophosphate (Sigma-Aldrich, USA) and 10-nM dexamethasone (Sigma-Aldrich, USA) for 14 d, and Alizarin red (Sigma-Aldrich, USA) staining was performed to determine the mineralization. For adipogenic differentiation, the seeded MSCs at the first passage were induced in media containing 0.5-mM isobutylmethylxanthine (MP Biomedicals, USA), 0.5-mM dexamethasone and 60-mM indomethacin (MP Biomedicals, USA) for 14 d, and Oil red O (Sigma-Aldrich, USA) staining was performed to determine the lipid droplet formation. Photographs were taken using an inverted optical microscope (CKX41; Olympus, Japan).
Culture of the 661W cell line was according to our previous study and verified for identity and non-contamination [70]. The 661W cell line was derived from mouse retinal tumors and has been characterized previously to be of cone photoreceptor cell lineage [71]. The 661W cell line was cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% FBS, 2-mM l-glutamine, 100-U/ml penicillin, and 100-g/ml streptomycin (all from Invitrogen, USA) at 37 °C in a humidified atmosphere of 5% CO2. For co-culture of MSCs with 661W cells, 661W cells were seeded in 12-well plates as the bottom of a Transwell system (0.4-μm pore size; Corning, USA), while MSCs at the first passage were added into the upper chamber of the Transwell system for 48 h.
Chemical treatments
MNU-provoked retinal degeneration model was established accordingly [70, 72] by intraperitoneal injection of MNU (Sigma-Aldrich, USA) at 40 mg/kg. MNU was freshly dissolved in sterile saline. Mice were sacrificed at indicated time points according to study design. In vitro treatment of 661W cells with MNU was performed at a concentration of 200 μg/ml for 6 h. For blockade of MSC generation of exosomes [36, 37], 10-μM GW4869 (Sigma-Aldrich, USA) was applied in MSC culture for 24 h, which was initially dissolved in DMSO into a stock solution of 5 mM and was diluted in culture media. The effects of GW4869 on MSC viability and osteogenesis were determined after wash-out procedures.
Collection and identification of exosomes
Collection of MSC-derived exosomes was performed as stated before [73]. Briefly, MSCs at nearly confluence of the first passage were cultured in exosome-depleted media (complete media supernatant after overnight centrifugation at 100,000 g) for 48 h. Exosomes from supernatants were then isolated by the differential centrifugation protocol at 4 °C: 300 g for 10 min, 3000 g for 10 min, 10,000 g for 20 min, 100,000 g for 70 min, followed by washing with PBS for another centrifugation at 100,000 g for 70 min. Quantification of exosomes were performed by determining the concentration of total proteins using the Pierce BCA Protein Assay (Thermo Fisher Scientific, USA).
Exosomes were identified according to the criteria of EVs [74]. The number and size distribution of exosomes was quantitated using dynamic light scattering with a NanosizerTM instrument (Malvern Instruments, UK) [75]. Transmission electron microscopy (TEM) [76] was performed on whole mounts of isolated exosomes using 2.5% glutaraldehyde, postfixed with 1% osmium tetroxide, embedded and stained with uranyl acetate and lead citrate, and observed using a TEM microscope (JEM-1230; JEOL, Japan). Purified exosomes were further characterized by western blot using anti-CD63 and anti-CD81 antibodies [73], as stated below.
Intravitreal injection
MSCs at the first passage were collected and suspended in PBS at 1 × 107 cells/ml and were injected at 1 μl into each vitreous chamber with a 33-G Hamilton syringe (Hamilton Company, USA) at 6 h after MNU treatment or at 2-week old of Pde6bmut mice. The collected exosomes were adjusted to a protein concentration of 1 μg/μl and were also injected at 1 μl into each vitreous chamber at 6 h after MNU treatment or at 2-week old of Pde6bmut mice. For the intravitreal delivery, the injection site was selected just posterior to the limbus and the needle was injected under anesthesia (100 mg/kg ketamine plus 20 mg/kg xylazine intraperitoneally), which was retracted after 2 min to minimize backflow [17].
Retinal histology and morphology
At sacrifice, retinal tissues were rapidly isolated, fixed overnight with 4% paraformaldehyde, and embedded in paraffin. 5-μm serial sections were prepared (RM2125; Leica, Germany) and underwent H&E staining for tissue histology and morphology, according to previous reports [70]. Quantification of ONL thickness was determined using the ImageJ 1.47 software along the vertical meridian of the eyeball from nasal to temporal side and through the optic nerve head (ONH) [77], and statistically analyzed as area under the curve (AUC).
TUNEL and IF staining
At sacrifice, retinal tissues were rapidly isolated, fixed in 4% paraformaldehyde, cryoprotected with 30% sucrose, and embedded in the optimal cutting temperature (OCT) compound. The specimens were snap-frozen and sectioned into 15-μm sagittal sections (CM1950; Leica, Germany). Sections were then underwent TUNEL assay using DeadEndTM Fluorometric TUNEL System according to the manufacturers’ instructions (Promega, USA), counterstained by Hoechst 33342 (Sigma-Aldrich, USA) [32]. The images were further analyzed using the ImageJ 1.47 software from at least five consecutive microscopic fields for TUNEL+ cells over total ONL cells. For IF analyses on cone photoreceptors, sections were blocked with 5% bovine serum albumin (BSA) (Sigma-Aldrich, USA) dissolved in PBS for 1 h at room temperature, and stained with a rabbit anti-mouse S-opsin primary antibody (ABN1660; Millipore, USA) overnight at 4 °C at a concentration of 1:1000. After washing with PBS, sections were stained with an AF647-conjugated goat-anti-rabbit secondary antibody for 1 h at room temperature, and were counterstained with Hoechst 33342 (Sigma-Aldrich, USA). Quantification of the percentages of positively stained area over total retinal fields was performed using the ImageJ 1.47 software.
ERG analysis
The UTAS Visual Diagnostic System with a Big Shot Ganzfeld (LKC Technologies, USA) was employed. Mice were dark-adapted overnight before ERG analysis. Under dim red light conditions, anesthesia was induced by an intraperitoneal injection of 80 mg/kg ketamine and 10 mg/kg chlorpromazine, and mice were then lightly secured to a stage to ensure a stable position for recording. Cornea was anesthetized with a drop of 0.5% proxymetacaine and was kept moist with physiological saline. Platinum circellus record electrodes were placed on each cornea, while reference and ground electrodes were respectively located in the mouth and inserted in the tail. White flashes with the intensity of 3.0 cd.s/m2 were applied for stimulating the responses. The band-pass (1–300 Hz) was used to amplify the recorded signals. The line noise was wiped off by a 50-Hz notch filter. Totally 10 scotopic (dark-adapted, mixed rod- and cone-mediated) and 60 photopic (light-adapted, cone-mediated) responses were recorded for analyzing the amplitudes of a-wave and/or b-wave by ERG View v4.380 R software [78].
Transfection of microRNA mimics, siRNA, and plasmids
The mimics and NC of miR-21, and the siRNA and NC of Pdcd4 were designed by RiboBio (Guangzhou, China) and were transfected into cells at final concentrations of 50 nM, according to previous studies [42, 43]. The Lipofectamine 2000 reagent (Invitrogen, USA) was used for transfection according to the manufacturer’s recommendation. CD63-EGFP plasmids were purchased from Hanheng (Shanghai, China) and were also transfected into MSCs using the Lipofectamine 2000 reagent (Invitrogen, USA). The transfected MSCs or 661W cells were further used for downstream experiments after 48 h incubation.
In vitro apoptosis assay
661W cells were harvested and evaluated by FITC-conjugated Annexin V and propidium iodide (PI) double staining according to the manufacturer’s instruction of Annexin V Apoptosis Detection Kit I (BD Biosciences, USA). Cell apoptosis was detected with a flow cytometer (CytoFLEX; Beckman Coulter, USA) equipped with the CXP 2.1 software. The percentages of early apoptotic (FITC+PI−) plus late apoptotic (FITC+PI+) cells were expressed as apoptotic percentages [55, 79].
Tracing of MSCs, exosomes, and miR-21
For tracing of MSCs and exosomes, PKH26 (Sigma-Aldrich, USA) was used according to manufacturer’s instructions [73]. For biodistribution of MSCs in the eye, freshly dissected eyes were scanned using the Xenogen IVIS imaging system (Xenogen IVIS Lumina II; PerkinElmer, USA) at 6 h after injection of PKH26-labeled MSCs. For in vivo tracing in the retina, retinal tissues were harvested at 24 h after injection of PKH26-labeled MSCs or exosomes. CD63-EGFP plasmid-transfected MSCs were also injected for in vivo examination of exosome release. For tracing of MSC-derived miR-21 in vivo, Cy5-labeled miR-21 mimics (RiboBio, China) was transfected into MSCs using the Lipofectamine 2000 reagent (Invitrogen, USA) before injection. The retina were then fixed in 4% paraformaldehyde, cryoprotected with 30% sucrose, and embedded in the OCT compound. The specimens were snap-frozen and sectioned into 15-μm sagittal sections (CM1950; Leica, Germany) and counterstained by Hoechst 33342 (Sigma-Aldrich, USA). For in vitro tracing, 661W cells were treated by PKH26-labeled exosomes for 48 h, fixed in 4% paraformaldehyde, blocked by 5% BSA, and immunostained with a rabbit anti-mouse primary antibody at a concentration of 1:100 for β-tubulin (ab18207; Abcam, UK) overnight at 4 °C. After washing with PBS, the sections were stained with a FITC-conjugated goat-anti-rabbit secondary antibody and counterstained by Hoechst 33342 (Sigma-Aldrich, USA). For tracing of MSC-derived miR-21 into 661W cells in vitro, Cy3-labeled miR-21 mimics (RiboBio, China) was transfected into MSCs using the Lipofectamine 2000 reagent (Invitrogen, USA) before MSCs being added in the upper chamber of the Transwell system. MSCs and 661W cells were then co-cultured for 48 h, and 661W cells were fixed in 4% paraformaldehyde and counterstained by Hoechst 33342 (Sigma-Aldrich, USA).
qRT-PCR
Total RNA was extracted from retinal tissues or cells by direct adding of Trizol Reagent (Takara, Japan) and purified by phenol–chloroform extraction. For mRNA, cDNA synthesis was performed using the PrimeScriptTM RT Reagent Kit (Takara, Japan), and the primer sequences of the genes detected in this study were listed in the Supplementary Information (Table S1). For microRNA, reverse transcription primers and qRT-PCR primers were designed by RiboBio (Guangzhou, China) as the Bulge-loopTM miRNA qRT-PCR Primer Sets. qRT-PCR detection was performed using the SYBR Premix Ex Taq II Kit (Takara, Japan) by a Real-Time System (CFX96; Bio-Rad, USA). The relative expression level of each gene was obtained by the cycle number after normalizing against Glyceraldehyde-3-phosphate dehydrogenase (Gapdh, for mRNA) and RNU6 (for microRNA) abundances using the 2−ΔΔCT method [42, 43].
Western blot
Traditional western blot was performed as previously described [79]. Whole lysates of exosomes were prepared using the Lysis Buffer (Beyotime, China). Proteins were extracted, loaded on sodium dodecyl sulfate-polyacrylamide gels, transferred to polyvinylidene fluoride membranes (Millipore, USA), and blocked with 5% BSA (Sigma-Aldrich, USA) in PBS with 0.1% Tween for 2 h in room temperature. The membranes were incubated overnight at 4 °C with the following primary antibodies: a rabbit anti-mouse primary antibody at a concentration of 1:1000 for CD63 (sc-5275; Santa Cruz Biotechnology, USA); and a rabbit anti-mouse primary antibody at a concentration of 1:1000 for CD81 (sc-70803; Santa Cruz Biotechnology, USA). The membranes were then incubated with peroxidase-conjugated goat anti-rabbit secondary antibodies at a concentration of 1:40000 for 1 h in room temperature. The blotted bands were visualized using an enhanced chemiluminescence Kit (Amersham Biosciences, USA) and a gel imaging system (5500; Tanon, China).
Capillary-based immunoassay was performed for retinal and 661W cell lysates using the Wes-Simple Western method with the anti-rabbit detection module (ProteinSimple, USA) [80]. Protein expression was measured by chemiluminescence and quantified as AUC using the Compass for Simple Western program (ProteinSimple, USA). Proteins were detected with the following primary antibodies: a rabbit anti-mouse primary antibody for Pdcd4 (9535S; Cell Signaling Technology, USA) and a rabbit anti-mouse primary antibody for Gapdh (5174S; Cell Signaling Technology, USA) at concentrations of 1:1000 (both from Cell Signaling Technology, USA).
Sample processing
Each experiment was performed in at least three independent times and data represented biological replicates. Sample size of animals was determined based on preliminary tests for variation to ensure adequate power for analysis. Samples were blindly allocated to each of the experimental group based on randomization and were also blind for data analysis. No samples were excluded from the results.
Statistical analysis
The results are represented as median ± range or as box (25th, 50th, and 75th percentiles) and whisker (range) plots for indicated experiments. The data were analyzed using non-parametric methods, i.e. the Mann–Whitney U tests for two group comparisons or the Kruskal–Wallis tests for multiple group comparisons, in the GraphPad Prism 5.01 software for not meeting normal distribution. Values of P < 0.05 were considered to be statistically significant.
References
Murakami Y, Notomi S, Hisatomi T, Nakazawa T, Ishibashi T, Miller JW, et al. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog Retin Eye Res. 2013;37:114–40.
Marigo V. Programmed cell death in retinal degeneration: targeting apoptosis in photoreceptors as potential therapy for retinal degeneration. Cell Cycle. 2007;6:652–5.
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86.
Park SS, Moisseiev E, Bauer G, Anderson JD, Grant MB, Zam A, et al. Advances in bone marrow stem cell therapy for retinal dysfunction. Prog Retin Eye Res. 2017;56:148–65.
Zakirova EY, Valeeva AN, Aimaletdinov AM, Nefedovskaya LV, Akhmetshin RF, Rutland CS, et al. Potential therapeutic application of mesenchymal stem cells in ophthalmology. Exp Eye Res. 2019;189:107863.
Jiang D, **ong G, Feng H, Zhang Z, Chen P, Yan B, et al. Donation of mitochondria by iPSC-derived mesenchymal stem cells protects retinal ganglion cells against mitochondrial complex I defect-induced degeneration. Theranostics. 2019;9:2395–410.
Millan-Rivero JE, Nadal-Nicolas FM, Garcia-Bernal D, Sobrado-Calvo P, Blanquer M, Moraleda JM, et al. Human Wharton’s jelly mesenchymal stem cells protect axotomized rat retinal ganglion cells via secretion of anti-inflammatory and neurotrophic factors. Sci Rep. 2018;8:16299.
Ezquer M, Urzua CA, Montecino S, Leal K, Conget P, Ezquer F. Intravitreal administration of multipotent mesenchymal stromal cells triggers a cytoprotective microenvironment in the retina of diabetic mice. Stem Cell Res Ther. 2016;7:42.
Bakondi B, Girman S, Lu B, Wang S. Multimodal delivery of isogenic mesenchymal stem cells yields synergistic protection from retinal degeneration and vision loss. Stem Cells Transl Med. 2017;6:444–57.
Arnhold S, Absenger Y, Klein H, Addicks K, Schraermeyer U. Transplantation of bone marrow-derived mesenchymal stem cells rescue photoreceptor cells in the dystrophic retina of the rhodopsin knockout mouse. Graefes Arch Clin Exp Ophthalmol. 2007;245:414–22.
Leow SN, Luu CD, Hairul Nizam MH, Mok PL, Ruhaslizan R, Wong HS, et al. Safety and efficacy of human Wharton’s jelly-derived mesenchymal stem cells therapy for retinal degeneration. PLoS ONE. 2015;10:e0128973.
Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis. 2008;14:782–806.
Reisenhofer M, Balmer J, Zulliger R, Enzmann V. Multiple programmed cell death pathways are involved in N-methyl-N-nitrosourea-induced photoreceptor degeneration. Graefes Arch Clin Exp Ophthalmol. 2015;253:721–31.
Sui BD, Hu CH, Liu AQ, Zheng CX, Xuan K, ** Y. Stem cell-based bone regeneration in diseased microenvironments: Challenges and solutions. Biomaterials. 2019;196:18–30.
Ranganath SH, Levy O, Inamdar MS, Karp JM. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell. 2012;10:244–58.
Jha KA, Pentecost M, Lenin R, Gentry J, Klaic L, Del Mar N, et al. TSG-6 in conditioned media from adipose mesenchymal stem cells protects against visual deficits in mild traumatic brain injury model through neurovascular modulation. Stem Cell Res Ther. 2019;10:318.
Mead B, Tomarev S. Bone marrow-derived mesenchymal stem cells-derived exosomes promote survival of retinal ganglion cells through miRNA-dependent mechanisms. Stem Cells Transl Med. 2017;6:1273–85.
Mathew B, Ravindran S, Liu X, Torres L, Chennakesavalu M, Huang CC, et al. Mesenchymal stem cell-derived extracellular vesicles and retinal ischemia-reperfusion. Biomaterials. 2019;197:146–60.
Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–79.
Zhou J, Benito-Martin A, Mighty J, Chang L, Ghoroghi S, Wu H, et al. Retinal progenitor cells release extracellular vesicles containing developmental transcription factors, microRNA and membrane proteins. Sci Rep. 2018;8:2823.
Mazzeo A, Lopatina T, Gai C, Trento M, Porta M, Beltramo E. Functional analysis of miR-21-3p, miR-30b-5p and miR-150-5p shuttled by extracellular vesicles from diabetic subjects reveals their association with diabetic retinopathy. Exp Eye Res. 2019;184:56–63.
Vidal-Gil L, Sancho-Pelluz J, Zrenner E, Oltra M, Sahaboglu A. Poly ADP ribosylation and extracellular vesicle activity in rod photoreceptor degeneration. Sci Rep. 2019;9:3758.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9.
Chen C, Wang D, Moshaverinia A, Liu D, Kou X, Yu W, et al. Mesenchymal stem cell transplantation in tight-skin mice identifies miR-151-5p as a therapeutic target for systemic sclerosis. Cell Res. 2017;27:559–77.
Wang B, Yao K, Huuskes BM, Shen HH, Zhuang J, Godson C, et al. Mesenchymal stem cells deliver exogenous MicroRNA-let7c via exosomes to attenuate renal fibrosis. Mol Ther. 2016;24:1290–301.
Sui BD, Zheng CX, Li M, ** Y, Hu CH. Epigenetic regulation of mesenchymal stem cell homeostasis. Trends Cell Biol. 2020;30:97–116.
Barbato S, Marrocco E, Intartaglia D, Pizzo M, Asteriti S, Naso F, et al. MiR-211 is essential for adult cone photoreceptor maintenance and visual function. Sci Rep. 2017;7:17004.
Ji HP, **ong Y, Song WT, Zhang ED, Gao ZL, Yao F, et al. MicroRNA-28 potentially regulates the photoreceptor lineage commitment of Muller glia-derived progenitors. Sci Rep. 2017;7:11374.
**ang L, Chen XJ, Wu KC, Zhang CJ, Zhou GH, Lv JN, et al. miR-183/96 plays a pivotal regulatory role in mouse photoreceptor maturation and maintenance. Proc Natl Acad Sci USA. 2017;114:6376–81.
Chu-Tan JA, Rutar M, Saxena K, Aggio-Bruce R, Essex RW, Valter K, et al. MicroRNA-124 dysregulation is associated with retinal inflammation and photoreceptor death in the degenerating retina. Investig Ophthalmol Vis Sci. 2018;59:4094–105.
Loscher CJ, Hokamp K, Wilson JH, Li T, Humphries P, Farrar GJ, et al. A common microRNA signature in mouse models of retinal degeneration. Exp Eye Res. 2008;87:529–34.
Sui B, Hu C, Zhang X, Zhao P, He T, Zhou C, et al. Allogeneic mesenchymal stem cell therapy promotes osteoblastogenesis and prevents glucocorticoid-induced osteoporosis. Stem Cells Transl Med. 2016;5:1238–46.
Sui BD, Chen J, Zhang XY, He T, Zhao P, Zheng CX, et al. Gender-independent efficacy of mesenchymal stem cell therapy in sex hormone-deficient bone loss via immunosuppression and resident stem cell recovery. Exp Mol Med. 2018;50:166.
Young B, Eggenberger E, Kaufman D. Current electrophysiology in ophthalmology: a review. Curr Opin Ophthalmol. 2012;23:497–505.
Rosolen SG, Kolomiets B, Varela O, Picaud S. Retinal electrophysiology for toxicology studies: applications and limits of ERG in animals and ex vivo recordings. Exp Toxicol Pathol. 2008;60:17–32.
Essandoh K, Yang L, Wang X, Huang W, Qin D, Hao J, et al. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochimica Biophysica Acta. 2015;1852:2362–71.
Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–7.
Yan W, Yao L, Liu W, Sun K, Zhang Z, Zhang L. A kind of rd1 mouse in C57BL/6J mice from crossing with a mutated Kunming mouse. Gene. 2017;607:9–15.
Li HJ, Sun ZL, Pan YB, Sun YY, Xu MH, Feng DF. Inhibition of miRNA-21 promotes retinal ganglion cell survival and visual function by modulating Muller cell gliosis after optic nerve crush. Exp Cell Res. 2019;375:10–19.
Han F, Huo Y, Huang CJ, Chen CL, Ye J. MicroRNA-30b promotes axon outgrowth of retinal ganglion cells by inhibiting Semaphorin3A expression. Brain Res. 2015;1611:65–73.
Zeng K, Wang Y, Yang N, Wang D, Li S, Ming J, et al. Resveratrol inhibits diabetic-induced muller cells apoptosis through MicroRNA-29b/specificity protein 1 pathway. Mol Neurobiol. 2017;54:4000–14.
Hu CH, Sui BD, Du FY, Shuai Y, Zheng CX, Zhao P, et al. miR-21 deficiency inhibits osteoclast function and prevents bone loss in mice. Sci Rep. 2017;7:43191.
Chen N, Sui BD, Hu CH, Cao J, Zheng CX, Hou R, et al. microRNA-21 contributes to orthodontic tooth movement. J Dent Res. 2016;95:1425–33.
Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–36.
Wei F, Yang S, Guo Q, Zhang X, Ren D, Lv T, et al. MicroRNA-21 regulates osteogenic differentiation of periodontal ligament stem cells by targeting Smad5. Sci Rep. 2017;7:16608.
Yang N, Wang G, Hu C, Shi Y, Liao L, Shi S, et al. Tumor necrosis factor alpha suppresses the mesenchymal stem cell osteogenesis promoter miR-21 in estrogen deficiency-induced osteoporosis. J Bone Min Res. 2013;28:559–73.
Zhao Q, Chen S, Zhu Z, Yu L, Ren Y, Jiang M, et al. miR-21 promotes EGF-induced pancreatic cancer cell proliferation by targeting Spry2. Cell Death Dis. 2018;9:1157.
Su W, Li Z, Jia Y, Zhu Y, Cai W, Wan P, et al. microRNA-21a-5p/PDCD4 axis regulates mesenchymal stem cell-induced neuroprotection in acute glaucoma. J Mol Cell Biol. 2017;9:289–301.
Scholl HP, Strauss RW, Singh MS, Dalkara D, Roska B, Picaud S, et al. Emerging therapies for inherited retinal degeneration. Sci Transl Med. 2016;8:368rv366.
Gagliardi G, Ben M’Barek K, Goureau O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: a pluripotent stem cell-based approach. Prog Retin Eye Res. 2019;71:1–25.
Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. 2015;385:509–16.
Bai L, Shao H, Wang H, Zhang Z, Su C, Dong L, et al. Effects of mesenchymal stem cell-derived exosomes on experimental autoimmune uveitis. Sci Rep. 2017;7:4323.
Oner A, Gonen ZB, Sinim N, Cetin M, Ozkul Y. Subretinal adipose tissue-derived mesenchymal stem cell implantation in advanced stage retinitis pigmentosa: a phase I clinical safety study. Stem Cell Res Ther. 2016;7:178.
Huang H, Kolibabka M, Eshwaran R, Chatterjee A, Schlotterer A, Willer H et al. Intravitreal injection of mesenchymal stem cells evokes retinal vascular damage in rats. FASEB J. 2019. https://doi.org/10.1096/fj.201901500R.
Zheng CX, Sui BD, Liu N, Hu CH, He T, Zhang XY, et al. Adipose mesenchymal stem cells from osteoporotic donors preserve functionality and modulate systemic inflammatory microenvironment in osteoporotic cytotherapy. Sci Rep. 2018;8:5215.
Mesentier-Louro LA, Zaverucha-do-Valle C, da Silva-Junior AJ, Nascimento-Dos-Santos G, Gubert F, de Figueiredo AB, et al. Distribution of mesenchymal stem cells and effects on neuronal survival and axon regeneration after optic nerve crush and cell therapy. PLoS ONE. 2014;9:e110722.
Vilela CAP, Souza LEB, Siqueira RC, Calado RT, Covas DT, Paula JS. Ex vivo evaluation of intravitreal mesenchymal stromal cell viability using bioluminescence imaging. Stem Cell Res Ther. 2018;9:155.
Tzameret A, Sher I, Belkin M, Treves AJ, Meir A, Nagler A, et al. Transplantation of human bone marrow mesenchymal stem cells as a thin subretinal layer ameliorates retinal degeneration in a rat model of retinal dystrophy. Exp Eye Res. 2014;118:135–44.
Huang C, Zhang J, Ao M, Li Y, Zhang C, Xu Y, et al. Combination of retinal pigment epithelium cell-conditioned medium and photoreceptor outer segments stimulate mesenchymal stem cell differentiation toward a functional retinal pigment epithelium cell phenotype. J Cell Biochem. 2012;113:590–8.
Shojaati G, Khandaker I, Funderburgh ML, Mann MM, Basu R, Stolz DB, et al. Mesenchymal stem cells reduce corneal fibrosis and inflammation via extracellular vesicle-mediated delivery of miRNA. Stem Cells Transl Med. 2019;8:1192–201.
Gutsaeva DR, Thounaojam M, Rajpurohit S, Powell FL, Martin PM, Goei S, et al. STAT3-mediated activation of miR-21 is involved in down-regulation of TIMP3 and neovascularization in the ischemic retina. Oncotarget. 2017;8:103568–80.
Shi L, Guo H, Li Z, Wang Y, Wang Y, Cui Y. Adenovirus-mediated down-regulation of miR-21-5p alleviates experimental autoimmune uveoretinitis in mice. Int Immunopharmacol. 2019;74:105698.
Gui F, Hong Z, You Z, Wu H, Zhang Y. MiR-21 inhibitor suppressed the progression of retinoblastoma via the modulation of PTEN/PI3K/AKT pathway. Cell Biol Int. 2016;40:1294–302.
Sims EK, Lakhter AJ, Anderson-Baucum E, Kono T, Tong X, Evans-Molina C. MicroRNA 21 targets BCL2 mRNA to increase apoptosis in rat and human beta cells. Diabetologia. 2017;60:1057–65.
William JNG, Wu J, Chen Z, Faruq O, Chang H. Overexpression of Mir-21-5p induces apoptosis and cell cycle arrest by down-regulating SKP2 and overcomes bortezomib resistance in multiple myeloma. Blood. 2019;134:1823.
Pan T, Jia P, Chen N, Fang Y, Liang Y, Guo M, et al. Delayed remote ischemic preconditioning confersrenoprotection against septic acute kidney injury via exosomal miR-21. Theranostics. 2019;9:405–23.
**ao J, Pan Y, Li XH, Yang XY, Feng YL, Tan HH, et al. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016;7:e2277.
Lv YJ, Yang Y, Sui BD, Hu CH, Zhao P, Liao L, et al. Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics. 2018;8:2387–406.
Sui B, Hu C, Liao L, Chen Y, Zhang X, Fu X, et al. Mesenchymal progenitors in osteopenias of diverse pathologies: differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci Rep. 2016;6:30186.
Hu CB, Sui BD, Wang BY, Li G, Hu CH, Zheng CX, et al. NDRG2 suppression as a molecular hallmark of photoreceptor-specific cell death in the mouse retina. Cell Death Discov. 2018;4:32.
Tan E, Ding XQ, Saadi A, Agarwal N, Naash MI, Al-Ubaidi MR. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Investig Ophthalmol Vis Sci. 2004;45:764–8.
Grimm C, Wenzel A, Behrens A, Hafezi F, Wagner EF, Reme CE. AP-1 mediated retinal photoreceptor apoptosis is independent of N-terminal phosphorylation of c-Jun. Cell Death Differ. 2001;8:859–67.
Liu S, Liu D, Chen C, Hamamura K, Moshaverinia A, Yang R, et al. MSC transplantation improves osteopenia via epigenetic regulation of notch signaling in lupus. Cell Metab. 2015;22:606–18.
Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, et al. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ Res. 2011;109:724–8.
Haga H, Yan IK, Takahashi K, Wood J, Zubair A, Patel T. Tumour cell-derived extracellular vesicles interact with mesenchymal stem cells to modulate the microenvironment and enhance cholangiocarcinoma growth. J Extracell Vesicles. 2015;4:24900.
Wang L, Li P, Tian Y, Li Z, Lian C, Ou Q, et al. Human umbilical cord mesenchymal stem cells: subpopulations and their difference in cell biology and effects on retinal degeneration in RCS rats. Curr Mol Med. 2017;17:421–35.
Tao Y, Chen T, Fang W, Yan Z, Yang Q, Huang Y, et al. The comparative efficiency of intraperitoneal and intravitreous injection of hydrogen rich saline against N-Methyl-N-Nitrosourea induced retinal degeneration: a topographic study. Front Pharm. 2017;8:587.
Sui BD, Hu CH, Zheng CX, Shuai Y, He XN, Gao PP, et al. Recipient glycemic micro-environments govern therapeutic effects of mesenchymal stem cell infusion on osteopenia. Theranostics. 2017;7:1225–44.
Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–6.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81870676 and 32000974), the Postdoctoral Innovative Talents Support Program of China (BX20190380), and the General Program of China Postdoctoral Science Foundation (2019M663986). We thank TC at Department of Clinical Medicine, Fourth Military Medical University for kindly donating of the Pde6bmut mice for the research.
Author information
Authors and Affiliations
Contributions
CLD, CBH, and STL contributed equal to the experimental performing, data acquisition and analysis, and manuscript drafting. NZ, LHB, and FZ contributed to data analysis and interpretation. YCX and TC contributed to data interpretation. BDS, XRY, and CHH contributed to the study conception and design, data interpretation and manuscript revision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Bianchi
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, CL., Hu, CB., Ling, ST. et al. Photoreceptor protection by mesenchymal stem cell transplantation identifies exosomal MiR-21 as a therapeutic for retinal degeneration. Cell Death Differ 28, 1041–1061 (2021). https://doi.org/10.1038/s41418-020-00636-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00636-4
- Springer Nature Limited