
Abstract
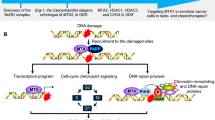
Herein, Tudor-SN was identified as a DNA damage response (DDR)-related protein that plays important roles in the early stage of DDR. X-ray or laser irradiation could evoke the accumulation of Tudor-SN to DNA damage sites in a poly(ADP-ribosyl)ation-dependent manner via interaction with PARP-1. Additionally, we illustrated that the SN domain of Tudor-SN mediated the association of these two proteins. The accumulated Tudor-SN further recruited SMARCA5 (ATP-dependent chromatin remodeller) and GCN5 (histone acetyltransferase) to DNA damage sites, resulting in chromatin relaxation, and consequently activating the ATM kinase and downstream DNA repair signalling pathways to promote cell survival. Consistently, the loss-of-function of Tudor-SN attenuated the enrichment of SMARCA5, GCN5 and acetylation of histone H3 (acH3) at DNA break sites and abolished chromatin relaxation; as a result, the cells exhibited DNA repair and cell survival deficiency. As Tudor-SN protein is highly expressed in different tumours, it is likely to be involved in the radioresistance of cancer treatment.
Similar content being viewed by others


Introduction
The integrity of genetic information is constantly subjected to exogenous threats, such as genotoxic agents, ionizing radiation (IR), ultraviolet radiation (UV) and endogenous changes [1, 2]. These threats induce an extensive range of DNA damage. Double-strand breaks (DSBs) are potentially the most harmful lesions, as they can eventually cause inflammation or tumourigenesis if the lesions are not promptly and correctly repaired [3, 4].
Upon encountering these threats, cells activate a DNA damage response (DDR), which is a sophisticated and coordinated process consisting of DNA lesion recognition, checkpoint activation, DNA repair and chromatin remodelling and apoptosis [5, 6]. One of the earliest events of DDR is the recruitment and activation of poly(ADP-ribose) polymerase-1 (PARP-1), which acts as a key molecular sensor of DNA breaks [7]. After activation, PARP-1 transfers ADP-ribose units from NAD+ to histones, acceptor proteins and PARP-1 itself, which is essential for initiating various forms of DDR pathways and facilitating DNA repair [7]. Either directly or via the recruitment of proteins, DNA damage-associated poly(ADP-ribosyl)ation (PAR), such as macroH2A1.1, the chromatin remodelling enzyme ALC1 and CHD4, induces the alteration of chromatin structures, resulting in chromatin relaxation and recruitment of other DDR proteins to the sites of DNA break [8,9,10]. Chromatin conformation is an essential event of DDR, in which DNA damage is efficiently accomplished [11, 12], as chromatin needs to undergo relaxation and be accessible to enable the accumulation of repair factors to the lesions. Although ATP-dependent chromatin remodelling and histone modifications are acknowledged for the modulation of chromatin structures in response to DNA damage [13], the underlying molecular mechanisms are sophisticated and remain elusive.
Tudor staphylococcal nuclease (Tudor-SN), which is also known as p100 or SND1 (staphylococcal nuclease domain containing 1), is implicated in a variety of cellular processes [Full size image
Poly(ADP-ribosyl)ation, which is catalysed by poly(ADP-ribose) polymerases (PARPs), has been demonstrated as a pivotal process of DDR to monitor DNA breaks and facilitate DNA repair [20]. To determine whether Tudor-SN is modified by PARP-1, MEF-WT cells were treated with or without PJ-34, which is a PARP-1 inhibitor. As shown in Fig. 1d, a very low level of poly(ADP-ribosyl)ated Tudor-SN was observed in untreated cells; however, it was significantly increased after exposure to IR. As expected, the addition of PJ-34 severely suppressed the DNA damage-induced poly(ADP-ribosyl)ation of Tudor-SN. These data suggest that Tudor-SN is modified by poly(ADP-ribosyl)ation in response to DNA damage.
A GST pulldown assay and co-immunoprecipitation assay were performed to map the specific domain of Tudor-SN that interacts with PARP-1. The bacterially produced GST-fusion proteins that contain full-length Tudor-SN (GST-FL), SN domain (GST-SN), TSN domain (GST-TSN) or GST alone were purified using glutathione-Sepharose beads. Equal amounts of fusion proteins were employed to bind assays with PARP-1 protein in MEF-WT cells. As shown in Fig. 1e, the full-length Tudor-SN and SN domain efficiently associated with PARP-1; the TSN domain was not effective. The physical interaction was determined in HeLa cells with ectopic overexpression of Flag-tagged full-length Tudor-SN (Flag-FL), the SN domain (Flag-SN), the TSN domain (Flag-TSN) or the control vector, as indicated. As shown in Fig. 1f, either the FL domain or the SN domain but not the TSN domain or negative ctrl associated with endogenous PARP-1. These data indicate that the SN domain of Tudor-SN mediates the interaction of these two proteins. To investigate whether SN domain can be modified by PARP-1, we ectopically overexpressed different Flag-tagged domains of Tudor-SN in HeLa cells. As shown in Fig. 1f, the SN domain and the full-length Tudor-SN were able to be poly(ADP-ribosyl)ated upon DNA damage. However, no detectable poly(ADP-ribosyl)ation of TSN domain was observed. Collectively, these data strongly support the idea that Tudor-SN is a potential substrate of PARP-1 that can be poly(ADP-ribosyl)ated in response to DNA damage.
DDR causes the accumulation of poly(ADP-ribosyl)ated Tudor-SN protein to the DNA damage sites
One of the hallmarks of DDR-related proteins is the local accumulation at DNA damage sites [21]. To investigate whether Tudor-SN is a bona fide protein that is involved in DNA damage, we performed a laser-induced DNA breaks assay to detect the accumulation of Tudor-SN at DNA damage sites. HeLa cells were transfected with GFP alone (ctrl) or ectopically overexpressed with GFP-tagged full-length Tudor-SN (GFP-FL) and then subjected to laser microirradiation. Analysis of GFP signal in the cells (Fig. 2a) indicated that GFP-tagged Tudor-SN was rapidly accumulated at irradiated sites. We further demonstrated that the SN domain (GFP-SN) was recruited to the damaged sites after laser exposure but the TSN domain (GFP-TSN) was not recruited (Fig. 2b). Poly(ADP-ribosyl)ation modification has been reported to play an important role in the recruitment of DDR-related proteins to DNA damage sites [22, 23]. Consistent with this concept, no detectable accumulation of SN domain or Tudor-SN was observed at the DNA lesion region with the addition of PJ-34 (Fig. 2c).

PARP-1 regulates Tudor-SN recruitment to DSBs. HeLa cells transiently transfected with GFP-FL (a), GFP-SN and GFP-TSN (b), were subjected to laser microirradiation. Representative fields were acquired at the indicated time points after microirradiation. Scale bar: 10 μm. c HeLa cells transiently transfected with GFP-FL and GFP-SN were subjected to laser microirradiation in the absence or presence of PJ-34 (10 μm). Scale bar: 10 μm. d ChIP was performed with anti-Tudor-SN or anti-IgG as a negative control in DR-GFP U2OS cells transfected with control vector or I-SceΙ. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; **P < 0.01 (n = 3). e Western blotting of HA-I-SceΙ
To verify that Tudor-SN can be recruited to the DNA damage sites, we took advantage of chromatin immunoprecipitation (ChIP) assay with U2OS-DR-GFP reporter cells [24, 25]. As shown in Fig. 2d, the ChIP assay revealed that Tudor-SN was enriched in the DNA damage sites. The overexpression of the endonuclease I-SceΙ was verified by western blotting (Fig. 2e). These data are consistent with previous results that reveal that DNA damage can enhance poly(ADP-ribosyl)ation modification of Tudor-SN, which is required for the efficient accumulation of Tudor-SN at the DNA damage sites, and demonstrate that Tudor-SN is a novel DDR-related protein.
Tudor-SN plays an important role in cell survival in response to DNA damage
To investigate the physiological function of Tudor-SN on the cellular response to radiation, we performed MTT and clonogenic cell survival assays to assess cell viability. As shown in Fig. 3a, b, the loss-of-function of Tudor-SN in MEF-KO cells exhibited a significant reduction in cell viability and survival in response to IR compared with MEF-WT cells. A statistical analysis (Fig. 3c) verified that the loss of Tudor-SN function sensitized cells to DNA damage. As shown in Fig. 3d, Tudor-SN was efficiently knocked out in MEF-KO cells. Supportively, knockout of Tudor-SN in HeLa (HeLa-KO) cells by CRISPR/Cas9 system strongly impeded colony formation (Fig. 3e). We performed rescue experiments in HeLa-KO cells. The colony formation assays indicate that ectopic overexpression of Tudor-SN, but not the negative vector control, can largely rescue the phenotype evoked by loss-of-function of Tudor-SN, which indicates that Tudor-SN is a key mediator of DNA damage-induced cell viability (Fig. 3e, f). The protein levels of Tudor-SN in different samples were verified by western blotting (Fig. 3g). Moreover, we employed flow cytometry to determine that the apoptosis rate of MEF-KO cells significantly increased with IR treatment in a time-dependent manner compared with MEF-WT cells (Fig. 3h, i). Collectively, these data demonstrate that Tudor-SN plays an important role in the evasion of cell death to promote cell survival.

Downregulation of Tudor-SN sensitizes cells to DNA damage. a Cell viability of MEF cells after IR. MEF-WT and MEF-KO cells were treated with the indicated doses of radiation and analysed using an MTT assay after 3 days in culture. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *, P < 0.05 (n = 3). b MEF-WT and MEF-KO cells were treated with increasing doses of radiation and evaluated by a colony-forming assay after 10 days of culture. c Quantification of survival rate. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *P < 0.05 (n = 3); **P < 0.01 (n = 3). d Expression level of Tudor-SN in MEF-WT and MEF-KO cells. e Colony formation assay. HeLa-KO cells were transfected with Flag-Tudor-SN or vector and irradiated with the indicated doses of IR. f Quantification of survival rate. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *, P < 0.05 (n = 3). g Western blotting of Tudor-SN. h Compared with MEF-WT cells (24 h: 10.83 ± 1.26%, 48 h: 14.37 ± 1.76%, 72 h: 25.67 ± 2.43%), the apoptosis rate of MEF-KO cells was significantly increased (24 h: 20.06 ± 1.54%, 48 h: 29.08 ± 2.44%, 72 h: 41.42 ± 3.08%) with 20 Gy IR treatment. i Quantification of apoptosis percentage. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *P < 0.05 (n = 3); **P < 0.01 (n = 3)
Tudor-SN accelerates chromatin relaxation in response to DNA damage
An early event in the DDR associated with post-translational poly(ADP-ribosyl)ation modification activity is the induction of chromatin relaxation, which enables DDR-related proteins to access the lesion sites [26]. Consistently, chromatin relaxation analysis showed that, in MEF-WT cells (lanes 6 and 7), but not MEF-KO cells with loss-of-function of Tudor-SN (lanes 9 and 10), the chromatin degraded into fragments with the addition of micrococcal nuclease (MNase) after IR treatment. The protein levels of Tudor-SN in different samples are shown in Fig. 4b. This finding demonstrates that Tudor-SN plays a crucial role in chromatin relaxation in response to DNA damage.

Tudor-SN mediates chromatin relaxation in response to DNA damage. a Chromatin relaxation assay was performed with different concentration of the nuclease (0, 0.5, or 0.8 U) to determine the extent of MNase sensitivity in MEF cells with 20 Gy IR. b Expression level of Tudor-SN in MEF-WT and MEF-KO cells. c, d Endogenous Tudor-SN associates with SMARCA5 in vivo. e, f Endogenous Tudor-SN associates with GCN5 in vivo. MEF-WT cells untreated or irradiated with 10 Gy IR. g Knockout of Tudor-SN reduces the acetylation level of H3K9, 14 and 18 upon DNA damage. h ChIP assay was performed with anti-Tudor-SN, anti-γH2AX, anti-acH3, anti-GCN5, anti-SMARCA5 or anti-IgG as a negative control in DR-GFP U2OS cells with or without I-SceΙ transfection. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; **P < 0.01 (n = 3). i−k ChIP of SMARCA5 (i), GCN5 (j) and acH3 (k) in DR-GFP U2OS cells transfected with Tudor-SN siRNA or scramble (Scr) siRNA in the absence or presence of I-SceΙ expression. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *P < 0.05 (n = 3); **P < 0.01 (n = 3). l Western blotting of Tudor-SN and HA-I-SceΙ
SMARCA5 ATPase, which was also identified in our mass spectrometry data, has been reported to be the catalytic subunit of the SWI/SNF family that contributes to remodel chromatin and facilitates the DNA repair process [25, 27]. Therefore, co-immunoprecipitation assays were performed to show that IR treatment significantly enhanced the association of endogenous Tudor-SN with SMARCA5 (Fig. 4c, d). Additionally, a cooperative activation between ATP-dependent chromatin remodelling complexes and GCN5 is required to facilitate DSB repair [28]. We have recently reported that GCN5, which directly implicates in DSB response, can interact with the Tudor-SN [29]. Thus, we performed co-immunoprecipitation assays to reveal that IR treatment significantly enhanced the interaction of endogenous Tudor-SN and GCN5 compared with the untreated cells (Fig. 4e, f).
GCN5 is a histone acetyltransferase that acetylates histone H3 on lysine 9, 14 and 18 (H3K9ac, H3K14ac and H3K18ac) [30, 31]. The acetylation of these marks is directly involved in the unfolding of high-order chromatin structures in the DDR; thus, we examined the different acetylation status of H3. As shown in Fig. 4g, the acetylation level of H3K9ac, H3K14ac and H3K18ac was significantly increased in an IR dose-dependent manner in MEF-WT cells; however, very low acetylation levels were observed in MEF-KO cells without an observed difference.
Furthermore, we investigate whether the Tudor-SN-dependent chromatin remodelling specifically occurs at DDR foci. We performed a ChIP assay in U2OS-DR-GFP reporter cells and discovered that the effective accumulation of Tudor-SN, γH2AX, acetylation of histone H3 (acH3), GCN5 and SMARCA5 coincided at the I-SceΙ-generated break sites (Fig. 4h). We subsequently performed a ChIP assay with anti-SMARCA5, anti-GCN5 and anti-acH3 in U2OS cells to verify that Tudor-SN is required for chromatin remodelling. As shown in Fig. 4i–k, the loss-of-function of Tudor-SN remarkably attenuates the enrichment of SMARCA5, GCN5 and acH3 at the defined DNA break sites compared with wild-type U2OS cells. The knockdown effect of Tudor-SN and the overexpression of the endonuclease I-SceΙ were verified by western blotting (Fig. 4l). These data indicate that Tudor-SN is involved in the early stage of DDR, which contributes to chromatin relaxation at DNA damage sites.
The activation of ATM signal pathway is remarkably inhibited due to un-sufficient chromatin relaxation impaired by loss-of-function of Tudor-SN protein
DNA damage-induced chromatin relaxation is required and can initiate ATM activation, which is crucial for the initiation of DDR signal pathways [32, 33]. To reveal the essential role of Tudor-SN as a regulator of chromatin remodelling in DDR, we detected the phosphorylation of a subset of ATM substrates, which were consequently activated by triggered ATM activation. As shown in Fig. 5a, IR treatment induced or enhanced the phosphorylation of ATM and downstream targets, including Chk2 and p53 in MEF-WT cells compared with MEF-KO cells.

Tudor-SN facilitates the DNA damage-induced phosphorylation of a subset of ATM substrates. a Total extracts from MEF-WT and MEF-KO cells were immunoblotted against the indicated antibodies following 10 Gy IR. b Expression level of γH2AX in MEF-WT and MEF-KO cells after the indicated doses (0, 2, 6 and 10 Gy) of IR. c The graph represents densitometric units normalized to H2AX. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; **P < 0.01 (n = 3). d γH2AX expression was detected by flow cytometry after the indicated doses of IR. e Quantification of γH2AX percentage. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *P < 0.05 (n = 3); **P < 0.01 (n = 3). f Immunofluorescence assay shows significantly reduced γH2AX foci formation in MEF-KO cells than MEF-WT cells after 10 Gy IR. Scale bar: 10 μm. g The quantification of γH2AX foci number per nucleus. Fifty nuclei were counted in each group. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; **P < 0.01 (n = 3). h Co-localization of endogenous Tudor-SN with γH2AX upon DNA damage. The distribution of endogenous Tudor-SN and γH2AX was detected using immunofluorescence with anti-Tudor-SN and anti-γH2AX by confocal microscopy analysis. Scale bar: 10 μm
Phosphorylation of histone variant H2AX (γH2AX) is a hallmark of DDR activation mediated by ATM and is essential for the accumulation of repair factors and amplification of the DNA damage signal [34]. Therefore, we employed different methodologies to examine the γH2AX level in MEF cells with loss-of-function of Tudor-SN. As shown in Fig. 5b, c, the protein level of γH2AX was significantly increased in MEF-WT cells in a dose-dependent manner with IR treatment but was rarely detectable in MEF-KO cells. Flow cytometry demonstrated an increased number of MEF-WT cells with γH2AX in a dose-dependent manner after IR treatment, but this was not observed in MEF-KO cells with impaired γH2AX levels (Fig. 5d, e). In addition, the immunofluorescence assay illustrated that MEF-WT cells displayed a significantly large number of γH2AX foci after IR, whereas less and weaker γH2AX foci formation was detected in MEF-KO cells for the same condition (Fig. 5f, g), which suggests a prominent role of Tudor-SN in the formation of γH2AX foci. Additionally, immunofluorescence assay demonstrated the co-localization of Tudor-SN and γH2AX in the foci of the cells with IR treatment (Fig. 5h). Collectively, these findings support the idea that less phosphorylation of ATM signal pathway in MEF-KO cells is not attributed to less DNA damage but rather is due to inefficient chromatin relaxation at DSBs, which is consistent with the observation that loss-of-function of Tudor-SN causes inefficient chromatin relaxation.
DNA repair deficiency occurs in the cells with loss-of-function of Tudor-SN
The functional consequences of ATM signal pathway phosphorylation include the activation of cell cycle checkpoints, including the G2/M checkpoint in response to IR [35]. To address the possible involvement of Tudor-SN in cell cycle arrest after DNA damage, we performed cell cycle analyses. As shown in Fig. 6a, b, IR induced the accumulation of MEF-WT cells in the G2/M phases of the cell cycle. This accumulation was not observed in the MEF-KO cells, which suggests that Tudor-SN is important for the regulation of the cell cycle arrest in the G2/M phases after DNA damage, which provides time for repair of the DNA lesion.

Tudor-SN depletion impairs G2/M arrest and DNA repair. a The cell cycle distribution was detected by flow cytometry after IR treatment. b The percentage of cells in each phase are shown in the histogram. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *P < 0.05 (n = 3); **P < 0.01 (n = 3). c Assessment of DNA repair by comet assay. The comet tails of MEF-KO cells (52.2 ± 4.07 μm) were longer than the comet tails of MEF-WT cells (23.5 ± 3.51 μm) after 10 Gy IR treatment. d Comet tail length was determined for 50 cells using Casplab-Comet Assay Software. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; **P < 0.01 (n = 3). e Expression level of Tudor-SN in MEF-WT and MEF-KO cells. f In the HR assay, with ectopic overexpression of I-SceI in the cells, approximately 4.42±0.55% of GFP-positive cells were observed in the control U2OS-Scr cells, whereas fewer GFP-positive cells (2.65±0.31%) appeared in the U2OS-siRNA cells. g Quantification of GFP-positive cell percentage in the HR assay. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *P < 0.05 (n = 3). h In the NHEJ assay, with ectopic overexpression of I-SceI, the percentage of GFP-positive cells (3.66±0.49%) in HEK 293-siRNA cells was lower than the percentage of GFP-positive cells (6.32±0.71%) in the HEK 293-Scr control group. i Quantification of GFP-positive cell percentage in the NHEJ assay. Error bars indicate the s.d. Statistical analysis was performed using a two-tailed unpaired Student’s ttest; *P < 0.05 (n = 3). j U2OS and HEK 293 cells were transfected with Tudor-SN siRNA or Scr siRNA, and the transfection efficiency was detected by western blotting
Next, we investigate the impact of the loss-of-function of Tudor-SN on DNA repair activity to address the essential function of Tudor-SN in the early events of DDR. We performed a comet assay, which is an extremely sensitive assay, to evaluate the occurrence of DNA repair in MEF cells. As shown in Fig. 6c, d, the comet tails of MEF-KO cells were, on average, approximately 2.2-fold longer than the comet tails of MEF-WT cells after IR treatment, which indicated that the loss-of-function of Tudor-SN in MEF-KO cells impaired the DNA repair ability. The protein level of Tudor-SN in different samples is shown in Fig. 6e.
We also performed HR and NHEJ assays, which are two major methods for repairing DSBs, to demonstrate the importance of Tudor-SN in activating the early stage of DDR. DSBs were induced by the transient expression of I-SceΙ in different cells. Once the DSB is repaired by HR or NHEJ, a functional GFP gene is generated. Analysis of HR and NHEJ assay (Fig. 6f–i) indicated that depletion of Tudor-SN expression was associated with a reduced percentage of GFP-positive cells to nearly 40% of that of control cells, suggesting that Tudor-SN is required for efficient DNA repair. The knockdown effect of Tudor-SN and the overexpression of the endonuclease I-SceΙ were verified by western blotting (Fig. 6j). These data are consistent with our findings that Tudor-SN plays an important role in regulating chromatin relaxation to enable the accumulation of repair factors to the lesions, efficiently accomplishing DNA damage repair to promote cell survival under genotoxic stress.

