
Abstract
Metabolic cardiomyopathy (MC) is characterized by intracellular lipid accumulation and utilizing fatty acids as a foremost energy source, thereby leading to excess oxidative stress and mitochondrial dysfunction. There is no effective therapy available yet. In this study we investigated whether defective mitophagy contributed to MC and whether urolithin A (UA), a naturally occurring microflora-derived metabolite, could protect against MC in experimental obese mice. Mice were fed high fat diet for 20 weeks to establish a diet-induced obese model. We showed that mitochondrial autophagy or mitophagy was significantly downregulated in the heart of experimental obese mice. UA (50 mg·kg−1·d−1, for 4 weeks) markedly activated mitophagy and ameliorated MC in obese mice by gavage. In PA-challenged H9C2 cardiomyocytes, UA (5 μM) significantly increased autophagosomes and decreased autolysosomes. Furthermore, UA administration rescued PINK1/Parkin-dependent mitophagy and relieved mitochondrial defects in the heart of obese mice, which led to improving cardiac diastolic function and ameliorating cardiac remodelling. In PA-challenged primarily isolated cardiomyocytes, both application of mitophagy inhibitor Mdivi-1 (15 μM) and silencing of mitophagy gene Parkin blunted the myocardial protective effect of UA. In summary, our data suggest that restoration of mitophagy with UA ameliorates symptoms of MC, which highlights a therapeutic potential of UA in the treatment of MC.
Similar content being viewed by others

Introduction
The prevalence of obesity is increasing rapidly, and obesity has gradually become an epidemic; more than six million adult individuals worldwide have obesity [1, 2]. Globally, over 40% of deaths associated with high body mass index (BMI) are attributable to cardiovascular disease among the obese population [1]. In the setting of obesity, patients develop insulin resistance and hyperlipidaemia, which cause the accumulation of lipid droplets (LDs) in the heart. Excess LD accumulation in the cytosol of cardiomyocytes leads to structural and functional disturbances in the heart, resulting in cardiac lipotoxicity and finally heart failure (HF) [3, 4]. Accumulating evidence suggests that patients with obesity are at high risk of develo** a metabolic cardiomyopathy (MC) phenotype, which is independent of coronary artery disease, hypertension, and other cardiac diseases [5, 6]. The prevalence of MC is anticipated to increase substantially in parallel with the epidemic of obesity;[7] however, the underlying mechanisms remain elusive, and breakthrough therapies are urgently needed.
Growing evidence has demonstrated that mitochondrial defects play a key role in the pathogenesis of MC in both preclinical models of obesity and human patients [5, 8,9,10]. In obesity, the heart manifests a cardiac phenotype characterized by left ventricular hypertrophy, LD accumulation and foetal gene reactivation, which together lead to cardiac dysfunction [6, 11]. Cardiomyocytes in obesity are typified by impaired mitochondrial respiration, altered organelle localization, and mitophagy disarrangement [9, 11]. Mitochondria undergo dynamic processes of fission, fusion, biogenesis and mitophagy, among which mitophagy acts as a key pathway for mitochondrial degradation and homeostasis [12, 13]. Mitophagy, as a highly conserved form of selective autophagy, is critical for the removal and recycling of damaged or long-lived mitochondria via the PINK1/Parkin- or mitophagy receptor-dependent pathway [13, 14] Obviously enlarged mitochondria with collapsed membrane potential are observed in the cardiomyocytes of mice with MC compared with control mice [9, 15]. These findings suggest that mitochondrial dysfunction might be triggered by the accumulation of injured mitochondria that are not appropriately cleared by mitophagy.
In the present study, we explored whether defective mitophagy is an indicator of MC. Moreover, we demonstrated that urolithin A (UA) enhanced PINK1/Parkin-dependent mitophagy in cardiomyocytes and in a mouse model of MC. UA is the most bioactive gut microbiota-produced metabolite of ellagitannins, which are a class of polyphenols enriched in pomegranates, walnuts, and berries [16]. The health benefits of UA and its potential applications are being investigated. Recently, Andreux et al. reported a first-in-humans clinical trial showing the safety of UA and its ability to improve mitochondrial and skeletal muscle health in humans [17]. However, the effects and the molecular mechanism of UA in obesity-induced cardiomyopathy remain elusive. Here, we found that UA alleviated diastolic dysfunction, adverse remodelling and mitochondrial defects in obese mice. Mitophagy is closely associated with mitochondrial function and is indispensable for mitochondrial homeostasis in health and multiple disease conditions [15, 18, 19]. We hypothesized that rescuing proper mitophagy in the heart might eliminate injured mitochondria, restock cells with efficient organelles, and mitigate MC cardiac dysfunction and remodelling.
Materials and methods
A detailed description of materials and methods is provided in the online Supplementary Materials and Methods.
Animals and high-fat diet feeding
Healthy male C57BL/6 mice, SPF grade, aged 6–8 weeks, with body weights of 20 ± 5 g were purchased from the Medical Experimental Animal Center of Guangdong Province. The mice were reared in separate cages with food and water available ad libitum. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH publication no. 8023, revised 1978). The protocol was approved by the Institutional Animal Care and Use Committee, Guangzhou Medical University, Guangzhou, China.
A total of 56 mice were randomized to 4 groups equally. The mice were fed a high-fat diet (HFD, 60 kcal% fat, Research Diets D12492) for 20 weeks and then subjected to UA (50 mg·kg−1·d−1 by gavage) or vehicle for another 4 weeks concurrent with continuous HFD feeding. The control mice were fed a standard chow diet (ND, 10 kcal% fat, Research Diets D12450J).
Cell culture
Neonatal rat cardiomyocytes (NRCMs) were isolated from Sprague–Dawley rats aged 1–3 postnatal days. The ventricles were isolated, cut into 1 mm3 pieces and digested with 0.05% trypsin solution (without EDTA) overnight at 4 °C. Then, the tissues were processed in Type II Collagenase solutions (0.2%) at 37 °C for 45 min with gentle shaking. Then, the lysates were filtered through a 75 μm cell strainer mesh 2 or 3 times until most of the tissue was digested. The cell lysates were centrifuged (800 × g) for 5 min, resuspended in medium (DMEM, C11995500BT) supplemented with 15% foetal bovine serum (FBS) and plated in 100 mm dishes for 1 h to remove attached fibroblasts. The residual supernatants with abundant cardiomyocytes were replated on collagen-coated culture dishes. After 24 h, the cardiomyocytes were washed, and the medium was replaced with fresh medium for further experiments, including cell surface area and mitochondrial function tests.
H9C2 cardiomyocytes were obtained from the American Type Culture Collection and cultured as described previously [20].
To examine the effect of UA on PA induced cell dysfunction, the cells were treated with BSA-conjugated palmitic acid (PA, 400 μM) for 24 h with or without UA (1, 5, 10 μM). UA at concentration of 5 μM was used if not mentioned thereafter for the in vitro experiment.
Statistical analysis
All the data were analysed with GraphPad Prism 8.0 (GraphPad Software) and are expressed as the means ± SEMs. Differences between two groups were analysed using Student’s t test. For three or more groups, the normal distribution was confirmed by the Shapiro–Wilk test (P > 0.1) with SPSS 17.0. Differences among three or more groups were analysed using one-way ANOVA. P < 0.05 was considered to indicate statistical significance. Further Tukey post hoc analysis (α = 0.05) was performed to confirm where the differences occurred between groups.
Results
Impaired mitophagy is a hallmark of metabolic cardiomyopathy
To explore the relevance of mitophagy in the setting of metabolic cardiomyopathy, we established a preclinical obesity model by feeding C57BL/6 mice a high-fat diet (HFD, 60 kcal %) for up to 20 weeks. The expression of several mitophagy genes, such as Pink1, Park2, Atg9, Mfn1 and Rab7, was decreased in the cardiac tissues of the obese mice compared with the control mice fed a standard chow diet (ND, 10 kcal% fat) (Fig. 1a). Moreover, TEM observation demonstrated an increased ratio of damaged mitochondria (characterized by deformation, swelling, cristae fracture and rupture) in the cardiomyocytes of the mice fed on HFD. The numbers of autophagosomes or autolysosomes containing mitochondrial fractions were decreased, confirming impaired mitophagy in the hearts of obese mice (Fig. 1b). Furthermore, defective mitophagy in the hearts of obese mice was evidenced by immunoblotting of phospho-S65-ubiquitin, an indicator of PINK1-dependent phosphorylation of ubiquitinated mitochondrial proteins [21], and LC3 and P62 of the mitochondrial fraction, direct markers of mitophagy activation, all of which were downregulated in cardiac tissues in the hearts of obese mice compared with those of control mice fed a normal diet (ND). These findings confirmed that mitophagy was defective in the hearts of obese mice (Fig. 1c–f).

a mRNA expression of mitophagy genes (Pink1, Park2, Atg9, Mfn1, Rab7 and Bnip3) in cardiac tissue from mice fed a normal diet (ND) or a high-fat diet (HFD) (n = 5 mice per group). b Representative TEM images of heart tissues (scale bar, 2 µm). Arrows indicate autophagosomes or autolysosomes containing the mitochondrial fraction. Arrowheads indicate damaged mitochondria. c Representative image and d quantification of immunoblotting using mitochondrial fractions against phospho-S65-ubiquitin and VDAC as a loading control in the cardiac tissues of mice on an ND or a HFD (n = 4). e Representative image and f quantification of immunoblotting using mitochondrial fractions against LC3, P62 and Parkin and VDAC as a loading control in the cardiac tissues of mice on an ND or a HFD (n = 4). Data are shown as the mean ± SEM; *P < 0.05, **P < 0.01, vs. ND; unpaired Student’s t test.
Urolithin A alleviated mitophagy defects in obese mice
Regarding the decline in mitophagy, we investigated whether UA, a natural compound which has been shown to be able to promote mitophagy in worms, cells and mammals [22,23,24], could alleviate the mitochondrial defects that occur in metabolic cardiomyopathy. UA treatment increased the expression of LC3 and P62 in the mitochondrial fraction of the cardiac tissues compared with that in the obese mice (Fig. 2a, b). Moreover, the levels of the mitophagy-regulated genes Parkin and PINK1 were increased in UA-treated obese mice (Fig. 2a, b). In addition, electron microscopy assay was performed to observe mitophagosomes. Numerous swollen mitochondria and a reduced matrix density were observed in the mice fed on HFD. In contrast, UA treatment significantly increased the occurrence of autophagic vesicles encasing damaged mitochondria and improved microstructural organization in the HFD-fed mice (Fig. 2c). Together, these data indicated that UA activated mitophagy in the hearts of obese mice.

a Representative images and b analysis results of immunoblotting against p62 and LC3 of the mitochondrial fraction and Parkin and Pink1 of the total lysate in the cardiac tissues of mice on a HFD after UA treatment (n = 4). c Representative TEM images of mitochondrial morphology (scale bar, 800 nm) are shown. Arrows indicate autophagosomes or autolysosomes containing the mitochondrial fraction. Arrowheads indicate damaged mitochondria. Data are shown as the mean ± SEM; *P < 0.05, **P < 0.01 vs. ND; #P < 0.05 vs. HFD; One-way ANOVA.
UA restored autophagy flux and mitophagy in vitro
We further observed the effect of UA on mitophagy activation in vitro. H9C2 cardiomyocytes were transfected with mRFP-GFP-LC3 adenoviruses and then treated with palmitic acid in the presence or absence of UA. mRFP-GFP-LC3 shows yellow fluorescence under basal conditions. When autophagosomes fuse with acidified lysosomes to form autolysosomes, mRFP-GFP-LC3 shows red fluorescence [25]. The results revealed visible accumulation of mRFP-GFP-LC3 (yellow, autophagosomes), increased numbers of GFP-LC3 dots and decreased levels of mRFP-LC3 (red, autolysosomes) in 24 h PA-challenged cardiomyocytes, which were alleviated by UA (Fig. 3a–c). Moreover, we transfected cardiomyocytes with Mito-Keima, which is an indicator of mitophagy that fluoresces green under basal conditions and red in acidified lysosomes. The numbers of Keima dots (ratio of 550/440) were decreased in PA-challenged cardiomyocytes, which was indicative of impaired mitophagy flux; however, this decrease was significantly attenuated by UA treatment (Fig. 3d, e). Along this line, we found that UA treatment significantly enhanced mitophagy, as revealed by the colocalization of GFP-LC3 and MitoTracker (Fig. 3f, g). In addition, the levels of the mitophagy markers LC3 and P62 in the mitochondrial fraction were both increased with UA treatment. PINK1 and Parkin levels were also increased (Fig. 3h, i). Of note, the LC3II level in the mitochondrial fraction was intensely increased upon treatment with an inhibitor of autophagy flux, bafilomycin A1 (a lysosomal V-ATPase inhibitor), under a normal environment. In response to 24 h PA challenge, bafilomycin A1 failed to increase mitochondrial LC3II levels, indicating blockade of mitophagy flux; however, this effect was nullified with UA (Fig. 3j, k).

a–c The increases in the numbers of autophagosomes and decreases in the numbers of autolysosomes in H9C2 cardiomyocytes following 24 h PA treatment were attenuated by UA treatment (n = 3 independent experiments). Representative images of mRFP-GFP-LC3 transfection are shown (scale bar, 30 µm). d Representative image of Mito-Keima transfection and e the fluorescence intensity ratio (550 nm/440 nm) following 24 h PA challenge with or without UA treatment are displayed (scale bar, 10 µm). f Representative image and g quantification of transfected GFP-LC3 adenovirus colocalization with MitoTracker (scale bar, 10 µm). h Representative images and (i) quantification of immunoblotting for p62 and LC3 in the mitochondrial fraction and Parkin and Pink1 in the total lysate of CMs after UA treatment (n = 4). j Representative image of immunoblotting and quantification k showing that PA challenge-induced suppression of mito-LC3II elevation (normalized to VDAC) in response to bafilomycin A1 (Baf, 100 nM) was compromised by UA treatment in CMs (n = 4). Data are shown as the mean ± SEM. (b, c, e, g, i) *P < 0.05, **P < 0.01, ***P < 0.001 vs. BSA + vehicle; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. PA + vehicle. (k) *P < 0.05 vs. the corresponding group without bafilomycin A1; #P < 0.05 vs. the BSA + vehicle + Baf group; &P < 0.05 vs. the PA + vehicle + Baf group. One-way ANOVA.
Together, these results suggested that UA treatment improved autophagy and mitophagy flux, which were suppressed in the obese condition both in vivo and in vitro.
UA treatment protected against diastolic dysfunction and cardiac remodelling in obese mice
Given that mitophagy is impaired in metabolic cardiomyopathy, we aimed to delineate the effect of UA, a mitophagy inducer, on metabolic cardiomyopathy. To this end, we first established a mouse obesity model by feeding C57BL/6 mice a high-fat diet (HFD, 60 kcal %) for up to 20 weeks. We found that mouse body weight and blood glucose levels were increased with HFD challenge (Supplementary Fig. 1a–c). Echocardiographic evaluation revealed no significant difference in left ventricular ejection fraction (LVEF) or left ventricular fractional shortening (LVFS) between obese and non-obese mice (Supplementary Fig. 2a, b). However, diastolic function, as assessed by pulsed-wave Doppler imaging, was decreased in the obese mice, as revealed by a decreased E/A ratio (Supplementary Fig. 2c). Along the same line, a well-established index of systolic-diastolic performance, the LV myocardial performance index, was impaired in the obese mice compared with the control mice (Supplementary Fig. 2d). Therefore, the mice were fed a HFD as described above or the ND as nondiabetic controls for 20 weeks and then treated with UA or vehicle for another 4 weeks concurrent with HFD feeding.
Transthoracic echocardiography was performed to determine cardiac function. As expected, a persistent preservation of LVEF and LVFS in both HFD-fed and ND-fed mice was observed (Fig. 4a–c). However, diastolic function, as assessed by pulsed-wave Doppler imaging, was decreased in the obese mice, as revealed by a decreased E/A ratio and an increased LV myocardial performance index (Fig. 4d–f). Remarkably, this detrimental LV diastolic dysfunction in the hearts of obese mice was attenuated with UA treatment, suggesting a protective role of UA against obesity-induced MC (Fig. 4a–f). Moreover, Masson’s trichome staining revealed that UA treatment significantly decreased cardiac fibrosis in the obese mice (Fig. 4g, h). In addition, the obese mice manifested cardiac hypertrophy, which was alleviated in the obese mice treated with UA, as evidenced by the heart weight ratio (Fig. 4i), cross-sectional area of cardiomyocytes (Fig. 4j, k), and mRNA level of prohypertrophic natriuretic peptide type A (Nppa) (Fig. 4l). Furthermore, the effect of UA treatment on glucose metabolism was observed. Unexpectedly, UA did not affect body weight, blood glucose level, oral glucose tolerance (OGTT) or insulin sensitivity (ITT) in the mice fed either the ND or HFD (Supplement Fig. 1a–e).

a Representative images of left ventricular M-mode echocardiographic tracing in short-axis view. b Percentage of left ventricular ejection fraction. c Fractional shortening of the left ventricle. d Representative pulsed-wave Doppler tracings. e Ratio between the mitral E wave and A wave (E/A). f Myocardial performance index. g Representative images of Masson’s trichrome staining of the left ventricle from mice in different experimental groups. Scale bars, 100 μm. h The percentage of fibrosis area was quantified according to the Masson’s trichrome-positive areas using NIH ImageJ. i Ratio of heart weight to body weight (HW/BW). j Representative images of heart sections stained with WGA. Scale bars, 50 μm. k Quantitative analysis of the cross-sectional area based on WGA staining. i mRNA expression level of atrial natriuretic peptide A (Nppa) in cardiac tissues of mice exposed to the given treatments as tested by real-time PCR. Data are shown as the mean ± SEM; n = 4–8 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. ND; #P < 0.05 vs. HFD; One-way ANOVA.
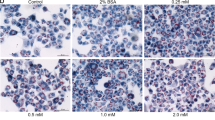
We further examined the effects of UA on cardiomyocyte hypertrophy. As expected, the cell surface areas of neonatal rat cardiomyocytes (NRCMs) were similar at the basal level. However, PA-induced cardiomyocyte hypertrophy was alleviated with UA treatment according to the cell surface area (Fig. 5a, b) and the mRNA levels of the established prohypertrophic genes Nppa and Nppb (Fig. 5c, d). Together, these results revealed that UA treatment protected against obesity-induced cardiomyopathy both in vivo and in vitro.

a NRCMs were subjected to palmitic acid treatment in the presence of different concentrations of UA. At the end of the experiment, the CMs were fixed in 4% paraformaldehyde and then stained with Alexa Fluor 568 phalloidin. A representative image is shown; n = 3 independent experiments. b Quantification of the surface area of cardiomyocytes; n = 50–70 cells per group. Scale bars, 50 μm. c mRNA expression levels of atrial natriuretic peptide A (Nppa) and d natriuretic peptide B (Nppb) in NRCMs exposed to the given treatments as determined by real-time PCR. Data are shown as the mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 vs. BSA + vehicle; ##P < 0.01, ###P < 0.001 vs. PA + vehicle. One-way ANOVA.
UA improved mitochondrial respiratory capacity and decreased lipotoxicity in vitro
To further explore the machinery whereby UA is protective against metabolic cardiomyopathy, we next investigated the UA-mediated effects on mitochondrial function. We used a Seahorse extracellular flux analyser to assess mitochondrial respiratory capacity in NRCMs. UA treatment elicited no discernible change under basal conditions (Fig. 6a, b). However, both the maximal and spare respiratory capacity were decreased in the PA-challenged cardiomyocytes, whereas these defects were alleviated by UA treatment (Fig. 6a–d). There was no visible difference in respiration based on ATP synthase or H+ leakage among all experimental groups (Fig. 6e, f). Moreover, an indicator of mitochondrial membrane potential (MMP), the JC-1 aggregate (red)/monomer (green) ratio, was tested in cardiomyocytes. The results showed that MMP was decreased in response to 24-h PA challenge, which was reversed by UA treatment (Fig. 6g, h). In addition, MitoSOX (red) intensely accumulated in response to 24 h PA challenge, whereas this accumulation was mitigated by UA treatment (Fig. 6i, j). UA alone exerted no visible changes in MMP and MitoSOX production under normal conditions. These results suggested that UA treatment conferred protection against mitochondrial dysfunction following PA challenge.

a Mitochondrial respiration in the NRCMs in response to PA challenge in the presence or absence of UA by measuring the cellular oxygen consumption rate (OCR). b Basal respiration, c maximal respiration, d spare respiration, e ATP synthesis and f H+ leak (n = 5). g Representative images and h analysis of JC-1 staining, scale bar: 30 μm. i Representative images of MitoSox staining in the cardiomyocytes, scale bar: 30 μm. j Quantification of the mitochondrial superoxide (MitoSox) positive intensity. Data were represented as the mean ± SEM, the experiment was repeated 3 times. **P < 0.01, ***P < 0.001 vs. BSA + vehicle; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. PA + Vehicle.
Mitophagy inhibition blunted the protective effect of urolithin A against cardiac lipotoxicity
To further explore the involvement of mitophagy flux in UA-mediated responses in cardiomyocytes in obesity, the mitophagy inhibitor Mdivi-1 was used. Our data revealed that UA-mediated myocardial protection was compromised by inhibition of mitophagy flux using Mdivi-1, which did not elicit more detrimental effects on PA-induced cardiomyocyte hypertrophy (Fig. 7a, b). Importantly, PA induced overt MitoSOX accumulation, which was attenuated by UA treatment; however, the effect was nullified by Mdivi-1 (Fig. 7c, d). The UA-mediated protection of MMP after PA challenge was also partially mitigated by Mdivi-1 (Fig. 7e).

a Representative image of NRCMs stained with Alexa Fluor 568 phalloidin, scale bar: 50 μm. b Quantification of surface area of cardiomyocytes in response to different treatments, as indicated; n = 50–70 cells per group. c Representative images of MitoSox staining in cardiomyocytes, scale bar: 30 μm. d The quantification of the MitoSox positive intensity. e Quantification of the of JC-1 staining. Data were represented as the means ± SEM. #P < 0.05, ###P < 0.001 vs. PA + vehicle. &&&P < 0.001 vs. PA + UA.
We further used genetic knockdown of the mitophagy gene parkin using siRNA to confirm whether UA mediated myocardial protection via mitophagy activation. As shown in Fig. 8, Parkin gene silencing significantly abrogated the protective effect of UA against PA challenge-induced cardiomyocyte hypertrophy, MitoSOX accumulation and MMP reduction. These findings suggested that mitophagy activation was involved in UA-mediated cardioprotective effects against obesity-induced MC.

a Representative image of NRCMs stained with Alexa Fluor 568 phalloidin, scale bar: 50 μm. b Quantification of surface area of cardiomyocytes in response to different treatments, as indicated; n = 50–70 cells per group. c Representative images of MitoSox staining, scale bar: 30 μm. d Quantification of the MitoSox positive intensity. e Quantification of the of JC-1 staining. Data were represented as the means ± SEM. ###P < 0.001 vs. PA + vehicle. &&P < 0.01, &&&P < 0.001 vs. PA + UA.
Discussion
The present study demonstrates that UA, a mitophagy inducer, protects against metabolic cardiomyopathy in experimental obesity. Several lines of evidence supported our conclusions. (i) Mitophagy was impaired in the heart in obesity. (ii) UA significantly activated mitophagy both in vivo and in vitro. (iii) UA protected against LV diastolic dysfunction and cardiac remodelling in obese mice without altering the blood glucose level. (iv) Mitochondrial defects due to PA challenge, including reduced respiratory capacity, MMP collapse and mitochondrial oxidative stress, were alleviated by UA treatment. (v) Either the pharmacological mitophagy inhibitor Mdivi-1 or silencing of the mitophagy gene Parkin blunted the effects of UA, confirming that UA-dependent myocardial protection against MC is attributable to mitophagy activation.
Our data demonstrate that defective mitophagy is a common feature of metabolic cardiomyopathy both in preclinical models of obesity and in vitro. It is still controversial whether mitophagy impairment is stringently associated with obesity and insulin resistance. However, several lines of evidence suggest that decreased mitochondrial energetics, reduced cardiac efficacy, shifted heart substrate usage, enhanced fatty acid oxidation and concurrent oxidative stress occur in both obese and diabetic patients [15, 26, 27]. An apparent bioenergetic impairment of cardiomyocyte mitochondria in diabetic cardiomyopathy is attributable to obesity in human patients [28]. A recent study has also suggested that mitophagy is related to obesity in a Parkin-dependent manner [29]. Along this line, Thomas et al. showed that Parkin, a critical mediator of mitophagy, is reduced in the heart in obesity, which indicates that Parkin-dependent mitophagy might underlie obesity-associated cardiovascular risk [30]. All of the above evidence, together with our results, suggests that obesity-associated cardiomyopathy is pathologically attributable to disruption of mitochondrial function homeostasis.
We showed mitophagy decline in the cardiac tissues of obese mice along with decreased cardiac diastolic performance, suggesting that deficient mitophagy contributes to metabolic cardiomyopathy. Therefore, we hypothesized that alleviation of mitophagy defects might restore mitochondrial quality and cardiac efficiency. To test our hypothesis, we treated obese mice with the mitophagy inducer UA, a naturally occurring microflora-derived metabolite. Our data demonstrated that UA enhanced Parkin-dependent mitophagy both in vivo and in vitro. In support of this notion, it has been revealed that UA stimulates mitophagy and improves muscle health in both experimental models and elderly humans [17, 22]. Moreover, we demonstrated that UA treatment significantly attenuated cardiac diastolic dysfunction and adverse remodelling in obese mice without altering body weight or blood glucose levels, suggesting a glucose-independent mechanism. In addition, both the pharmacological mitophagy inhibitor Mdivi-1 and silencing of the mitophagy gene Parkin blunted the UA-mediated cardiomyocyte protective effect in vitro, indicating the involvement of Parkin-dependent mitophagy in the protective effect of UA. In support of this notion, recent strategies to boost mitophagy have been revealed to improve cardiac performance in obese mice [29, 31]. Our previous study provided evidence that urolithin B, another bioactive gut metabolite of ellagitannins, protected the myocardium in a preclinical model of heart ischaemia/reperfusion injury [20]. Similarly, the effects of UA on both recycling of dysfunctional mitochondria and regeneration of new organelles were in line with previous data on UA in the context of aging [22, 32].
Mitophagy is a highly conserved selective autophagy process by which damaged mitochondria are degraded and replenish the cell to support mitochondrial homeostasis [12, 19]. Here, our results revealed that mitophagy was decreased in the cardiac tissues of obese mice. The mitophagy indicator Mito-Keima, under bafilomycin A1 treatment, directly indicated that mitophagy flux was impaired according to multiple parameters in the heart tissues of the obese mice. Our present data, together with the findings of a recent study [9], confirmed that impaired mitophagy was a hallmark of obesity-induced MC that could be attributable to the diminished efficiency of degradation and/or increased production of autophagosomes. However, the precise efficiency of digestion and the single determinant that is most important need more comprehensive investigation. Using immunoblotting of mito-LC3II and P62 in the mitochondrial fraction, MitoTracker and GFP-LC3 colocalization, and Mito-Keima staining, we detected that UA promoted mitophagy activation and mitophagy flux both in vivo and in vitro. Consistent with our findings, the effect of UA on mitophagy has been confirmed in multiple tissues, including skeletal muscle and adipocytes, by different groups [22, 24], which have also demonstrated that UA activates the Parkin mitophagy pathway. We cannot conclude whether UA targets Parkin directly or induces Parkin recruitment to damaged mitochondria via an indirect mechanism. Further investigation of the molecular mechanism by which UA regulates Parkin is warranted in the future.
Furthermore, we demonstrated that UA alleviated multiple mitochondrial defects in cardiomyocytes upon PA challenge, including overt mitochondrial oxidative stress, reduced mitochondrial respiratory capacity and MMP collapse. These effects might be attributable to UA-mediated activation of mitophagy, as revealed by the evidence that mitophagy activation contributed to the mitochondria-protective effect of UA. In turn, the improved mitochondrial quality could have helped alleviate the mitophagy impairment.
In summary, our present study suggests that UA treatment protects against diastolic dysfunction and cardiac remodelling, as well as mitochondrial defects, in the hearts of mice fed on HFD. The protective effects of UA are at least partially dependent on mitophagy activation. Thus, treatment to promote mitophagy may be a promising strategy to combat cardiac complications in obese patients.
References
Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, et al. Health effects of overweight and obesity in 195 countries over 25 Years. N Engl J Med. 2017;377:13–27.
Piché ME, Tchernof A, Després JP. Obesity phenotypes, diabetes, and cardiovascular diseases. Circ Res. 2020;126:1477–500.
Peterson LR, Gropler RJ. Metabolic and molecular imaging of the diabetic cardiomyopathy. Circ Res. 2020;126:1628–45.
Aminian A, Wilson R, Zajichek A, Tu C, Wolski KE, Schauer PR, et al. Cardiovascular outcomes in patients with type 2 diabetes and obesity: Comparison of gastric bypass, sleeve gastrectomy, and usual care. Diabetes Care. 2021;44:2552–63.
Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–38.
Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. 2017;136:6–19.
Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119.
Ren J, Wu NN, Wang S, Sowers JR, Zhang Y. Obesity cardiomyopathy: Eevidence, mechanisms, and therapeutic implications. Physiol Rev. 2021;101:1745–807.
Ren J, Sun M, Zhou H, Ajoolabady A, Zhou Y, Tao J, et al. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv. 2020;6:eabc8561. https://doi.org/10.1126/sciadv.abc8561.
Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitus. Circ Res. 2019;124:121–41.
Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res. 2016;118:1736–51.
Shirihai OS, Song M, Dorn GW 2nd. How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835–49.
Morales PE, Arias-Durán C, Ávalos-Guajardo Y, Aedo G, Verdejo HE, Parra V, et al. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Asp Med. 2020;71:100822.
Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015;116:1477–90.
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71.
Espín JC, Larrosa M, García-Conesa MT, Tomás-Barberán F. Biological significance of urolithins, the gut microbial ellagic Acid-derived metabolites: the evidence so far. Evid Based Complement Altern Med. 2013;2013:270418.
Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1:595–603.
Sorrentino V, Menzies KJ, Auwerx J. Repairing mitochondrial dysfunction in disease. Annu Rev Pharmacol Toxicol. 2018;58:353–89.
Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–8.
Zheng D, Liu Z, Zhou Y, Hou N, Yan W, Qin Y, et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol Res. 2020;153:104655.
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–6.
Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22:879–88.
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401–12.
Luan P, D’Amico D, Andreux PA, Laurila PP, Wohlwend M, Li H, et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci Transl Med. 2021;13:eabb0319. https://doi.org/10.1126/scitranslmed.abb0319.
Wu X, Qin Y, Zhu X, Liu D, Chen F, Xu S, et al. Increased expression of DRAM1 confers myocardial protection against ischemia via restoring autophagy flux. J Mol Cell Cardiol. 2018;124:70–82.
Peterson LR, Gropler RJ. Radionuclide imaging of myocardial metabolism. Circ Cardiovasc Imaging. 2010;3:211–22.
Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–66.
Niemann B, Chen Y, Teschner M, Li L, Silber RE, Rohrbach S. Obesity induces signs of premature cardiac aging in younger patients: the role of mitochondria. J Am Coll Cardiol. 2011;57:577–85.
Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124:1360–71.
Thomas A, Marek-Iannucci S, Tucker KC, Andres AM, Gottlieb RA. Decrease of cardiac parkin protein in obese mice. Front Cardiovasc Med. 2019;6:191.
Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu YL, et al. Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: Role of SIRT6. J Pineal Res. 2021;70:e12698.
D’Amico D, Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J. Impact of the natural compound urolithin a on health, disease, and aging. Trends Mol Med. 2021;27:687–99.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China [81573429 to Dr. X Wu; U1601227 to Dr. Yu], and Natural Science Foundation of Guangdong Province [2021A1515012149 to Dr. X Wu].
Author information
Authors and Affiliations
Contributions
XQW and XYY conceived the research. XQW and JRH designed the experiments. JRH, MHZ, YJC, YLS, ZMG, ZJL, GPZ and YQ conducted experiments. XQW and JRH drafted the manuscript. XYY and XYD made critical revision of the manuscript for key intellectual content. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary Material
Rights and permissions
About this article

Cite this article
Huang, Jr., Zhang, Mh., Chen, Yj. et al. Urolithin A ameliorates obesity-induced metabolic cardiomyopathy in mice via mitophagy activation. Acta Pharmacol Sin 44, 321–331 (2023). https://doi.org/10.1038/s41401-022-00919-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00919-1
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Mitochondrial quality control in human health and disease
Military Medical Research (2024)