
Abstract
As an important second messenger, calcium (Ca2+) regulates a wide variety of physiological processes. Disturbance of intracellular calcium homeostasis implicated in the occurrence of multiple types of diseases. Orai1 is the major player in mediating store-operated calcium entry (SOCE) and regulates calcium homeostasis in non-excitable cells. Over-expression and activation of Orai1 have been reported in breast cancer. However, its molecular mechanisms are still not very clear. Here, we demonstrated that Nucleolin (NCL) was a novel interacting partner of Orai1. NCL is a multifunctional nucleocytoplasmic protein and is upregulated in human breast tumors. The binding of C-termini of NCL (NCL-CT) to N-termini of Orai1 (Orai1-NT) is critical for mediating calcium influx and proliferation of breast cancer cells. Blocking the NCL-Orai1 interaction by synthesized Orai1 peptide can effectively reduce the intracellular calcium influx and suppress the proliferation of breast cancer cells in vitro and in vivo. Our findings reveal a novel activation mechanism of Orai1 via direct interaction with NCL, which may lead to calcium homeostasis imbalance and promote the proliferation of breast cancer cells. Blocking NCL-Orai1 interaction might be an effective treatment of breast cancer.
Similar content being viewed by others


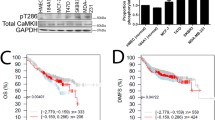
Introduction
Breast cancer is one of the most frequent malignancies in women and seriously threatens women’s health [1]. The pathogenesis of breast cancer is quite complicated. Accumulating evidence has demonstrated that calcium homeostasis imbalance and abnormal calcium signal are closely associated with the development of breast cancer [2,3,4]. Calcium, acting as the crucial second messenger, regulates multiple biological processes, including cell proliferation, migration, apoptosis, and gene transcription [5, 6]. So, intracellular calcium concentration is strictly regulated by membrane calcium channels, calcium pumps, and calcium-binding proteins.
SOCE is one of the main pathways of extracellular calcium influx in mammals, as well as the central mechanism of calcium signal regulation in non-excitable cells [7, 8]. It has been reported that augmented SOCE plays pivotal roles in regulating proliferation, invasion and metastasis of cancer cell [9,10,11]. SOCE is activated by depletion of intracellular Ca2+ stores and mediated by the interaction between the endoplasmic reticulum (ER)-located stromal interaction molecule 1 (STIM1) protein and the plasma membrane (PM)-embedded Orai1 at ER-PM junctions [12,13,14]. Dysfunction of STIM1/Orai1-mediated SOCE has been found in various tumor types, including breast cancer [3A). Because of containing multiple proteolytic cleavage sites in the N-terminal, full-length NCL is difficult to purify. Therefore, the NT truncated GST-NCL (284–710) fragment and GST proteins were expressed and purified from Escherichia coli, then incubated with Orai1-NT peptides immobilized to the streptavidin-conjugated beads. The bound proteins were analyzed by SDS-PAGE and coomassie staining, which showed an obvious GST-NCL protein precipitated by Orai1-NT (Fig. 3B, lane 4), whereas no precipitation of GST was observed (lane 3) and no binding was observed for beads alone (lane 5,6). Next, we mapped the domain of NCL responsible for interaction with Orai1-NT, whereas the central domain (284–629) of NCL was also difficult to purify. We observed that the purified NCL-CT fragment (630–710) was pulled down by the Orai1-NT peptide (Fig. 3C, lane 4). To further confirm the interaction domain, Flag-tagged full- length NCL and NCL fragments were expressed in MCF-7 cells. Cell lysate was incubated with biotin-Orai1-NT peptide followed by pulling-down with streptavidin beads and western blotting with anti-Flag antibody. Consistently, full-length NCL, NCL-ΔNT (284–710) and NCL-CT (630–710) exhibited interaction with Orai1 (Fig. 3D, lane 1,4, 5, left panel). Unexpectedly, NCL-NT (1–283) binding with Orai1 was also observed (Fig. 3D, lane 2, left panel). The whole cell lysates and bound peptides on the beads were revealed by western blotting and Coomassie staining (Fig. 3D, middle and right panels). Next, we tested the co-localization of NCL fragments with Orai1. MCF-7 cells were co-transfected with GFP-Orai1 and Flag-NCL fragment plasmids, then treated with or without TG, a blocker of sarcoendoplasmic reticulum calcium ATPase (SERCA) pump to induce SOCE, followed by immunofluorescence staining with anti-Flag antibody (red fluorescence). Expression of GFP-Orai1 exhibited its localization in the plasma membrane and NCL (1–283) mainly localized in the nucleoplasm. No obvious Orai1/NCL (1–283) co-localization was observed in the plasma membrane, whereas treatment with TG caused the accumulation of NCL (1–283) in the cytoplasm and plasma membrane which strongly co-localized with Orai1 (Fig. 3E). In addition, NCL (630–710) showed a clear shift in localization towards the cytoplasm and plasma membrane and exhibited strong co-localization with Orai1 in the plasma membrane with or without TG treatment (Fig. 3F). NCL (284–629) was found to localize in the nucleus, where no obvious Orai1/NCL co-localization was observed and no difference in co-localization was observed when cells were treated by TG (Fig. 3G). Furthermore, colocalization in the PM between NCL fragments and Orai1 was quantified (Fig. 3H). Thus, these results are consistent with the pull-down assay and suggest that both the NT and CT of NCL contributed to the interaction with Orai1.

A Schematic structure of NCL protein. B Biotin-based peptide pulldown assay to determine Orai1-binding domain of NCL. Purified GST and GST-NCL (284–710) proteins were incubated with the immobilized Orai1-NT peptides, then the precipitated samples were analyzed by SDS-PAGE. C Biotin-Orai1 peptide pulldown assay with different fragments of GST-NCL. D As in B, but with the MCF-7 cell lysate transfected with different NCL-Flag plasmids. The precipitated samples and cell lysates were detected by western blotting and bound peptides were analyzed by Coomassie staining. E Co-localization of GFP-Orai1 (green) with NCL (1–283)-Flag (red). MCF-7 cells co-transfected with GFP-Orai1 and NCL (1–283)-Flag plasmids were treated with or without TG. The cellular co-localization of Orai1 and NCL was detected by confocal microscopy. The boxed area was shown at high magnification in the panels on the right. F As in E, but co-localization of GFP-Orai1 with NCL (630–710)-Flag was assayed. G As in E, but co-localization of GFP-Orai1 with NCL (284–629)-Flag was assayed. H Quantification of colocalization between the fluorescent channels of NCL-Flag fragments and GFP-Orai1.
NCL-Orai1 interaction meditated store-operated calcium entry
To study the role of NCL-Orai1 interaction in the progression of breast cancer, we tested whether NCL was involved in SOCE. Firstly, we examined the effect of NCL expression level on TG-evoked SOCE in HEK293T cells by calcium imaging. Ca2+ influx was measured by monitoring the cytosolic Ca2+ levels with Ca2+ indicator Fura-2 AM after store depletion induced by TG. We found that when overexpression of GFP-NCL, SOCE was significantly elevated following addition of 2 mM Ca2+ compared with GFP control (Fig. 4A). Conversely, knocking down NCL expression by siRNA resulted in a decrease in SOCE compared with the control siRNA group (Fig. 4B, C). We next tested whether NCL regulated SOCE by interaction with Orai1. We also visualized the real-time alterations in the intracellular Ca2+ concentration in breast cancer cells through confocal microscopy in conjunction with red fluorescent Ca2+ indicator CalbryteTM 630 AM. Consistently, overexpression of GFP-NCL in MCF-7 cells and MDA-MB-231 cells significantly increased basal Ca2+ concentration and TG induced SOCE (Fig. S2). Therefore, our data suggested that NCL was implicated in the regulation of SOCE. We next tested whether NCL regulated SOCE by interaction with Orai1. Firstly, MCF-7 cells were co-transfected with 4TO-NCL and GFP-Orai1 WT or R91W mutant plasmids, then TG-evoked SOCE was measured. We observed that when NCL and Orai1 WT were co-expressed, SOCE was obviously elevated, but not when NCL and channel dead mutant R91W were co-expressed (Fig. 4D). Then we tested whether Orai1 channel inhibitor 2-APB affected NCL-meditated SOCE. As expected, the SOCE of GFP-NCL over-expressed cells was reduced after treatment with 2-APB (Fig. 4E). Additionally, we observed a dramatic decrease of TG-evoked SOCE when we depleted NCL using siRNA in cells overexpressing GFP-Orai1 (Fig. S3). Thus, our findings indicated that NCL played a crucial role in the regulation of SOCE via Orai1. To further elucidate the activation mechanism of Orai1 by NCL, the effects of NCL fragment overexpression on SOCE were investigated. We noticed that the N-terminus of NCL did not induce the calcium entry despite being able to interact directly with Orai1, and the central region of NCL did not enhance the SOCE as expected, whereas C-terminus was sufficient to increase basal Ca2+ concentration and induce SOCE (Fig. 4F). Next, we tested whether STIM1 is required for NCL-meditated activation of Orai1. STIM1 was knocked down by siRNA in MCF-7 cells (Fig. S4). Depletion of STIM1 did not result in a marked decrease of NCL-meditated SOCE suggesting that activation of Orai1 by NCL was independent of STIM1 (Fig. 4G). Taken together, our results suggested that the C-terminus of NCL interacts with Orai1 to activate Ca2+ influx, which may alter calcium-regulated signal and contribute to breast cancer progression.

A Representative traces of fura-2 Ca2+-imaging experiments in HEK293T cells transfected with GFP or GFP-NCL. Calcium stores were depleted with 1 μM TG, and SOCE was measured by the addition of 2 mM Ca2+ in the medium. B As in A, but HEK293T cells transfected with control siRNA or NCL siRNA. C Representative Fura-2 fluorescent ratio in HEK293T cells expressing GFP vector, GFP-NCL, siCtrl and siNCL. ***P < 0.001. D Live-cell imaging of intracellular calcium flux using confocal microscopy. MCF-7 cells co-transfected with 4TO-NCL and GFP-Orai1 WT or GFP-Orai1 R91W plasmids were loaded with red-fluorescent calcium indicator CalbryteTM 630 AM and imaged via laser scanning fluorescence confocal microscopy. After treatment with TG, the real-time alterations in intracellular Ca2+ concentration was measured. The calcium level in MCF-7 cells was expressed as the relative fluorescence intensity based on the fluorescence images. E As in D, but cells transfected with GFP-NCL were treated with or without 2-APB, then intracellular calcium flux was measured by confocal microscopy. F As in D, but cells were co-transfected with GFP-Orai1 and different fragments of 4TO-NCL plasmids. The real-time alterations in intracellular Ca2+ concentration was measured by confocal microscopy. G As in D, but MCF-7 cells transfected with GFP or GFP-NCL, then were transfected with STIM1 siRNA. After treatment with TG, the real-time alterations in intracellular Ca2+ concentration was measured. All data are shown as mean ± SD. of three replicates. *P < 0.05. **P < 0.01. ***P < 0.001.
NCL activates Orai1 to promote proliferation of breast cancer cells
To probe the linkage between NCL-meditated SOCE and the proliferation of breast cancer cells, we established the stable breast cancer cell lines (MCF-7 and MDA-MB-231) with constitutive overexpression of NCL and performed MTT assay (Fig. 5A), colony formation (Fig. 5B) and EdU proliferation assays (Fig. 5C, D). These assays revealed that NCL overexpression promoted proliferation and growth of breast cancer cells, but treatment with Orai1 channel blocker 2-APB abolished the promoting effects of NCL on cell proliferation and clonogenicity. In addition, wound healing assay showed the promoting effect of NCL on cell migration, but inhibition of SOCE with 2-APB significantly decreased migration ability that was promoted by NCL (Fig. 5E, F). A similar inhibition effect of 2-APB on NCL-meditated proliferation and migration was observed in MDA-MB-231 cells (Fig. S5). Our results suggested that NCL promoted the proliferation of breast cancer cells by increasing calcium influx.

A MTT assay assessed the effect of 2-APB on the proliferation of MCF-7 cells stable expression of NCL. MCF-7 cells stable expression of NCL and negative control cells were treated with different concentration of 2-APB, and then the viability of cells was evaluated. B Colony formation assay of MCF-7 cells stable expression of NCL and negative control cells treated with or without 2-APB. C EdU staining for evaluation of the influences of 2-APB on the proliferation of NCL-overexpression MCF-7 cells. Dividing cells were labeled with EdU (green). All cells were counterstained with DAPI (blue). D Quantitative analyses of the percentages of EdU-positive cells. E Scratch assay to evaluate migration ability of MCF-7 cells stable expression of NCL and negative control cells. After cell growth reached confluence, a scratch was made by a 200 μL pipette tip, and cells were treated with or without 2-APB for 24 h. The closure of the scratch was imaged and analyzed. F Wound closure rates were expressed as percentages of the wound area closed at 24 h relative to the initial area at 0 h. All data are shown as mean ± SD. of three replicates. *P < 0.05. **P < 0.01. ***P < 0.001.
Blocking the interaction between NCL and Orai1 with synthesized Orai1 peptide inhibited breast cancer cells proliferation
NCL-meditated SOCE was required for the proliferation of breast cancer cells, so blocking the binding between NCL and Orai1-NT may lead to the reduction in calcium influx and inhibition of cancer cells proliferation. To investigate this possibility, the Rhodamine-labeled TAT-Orai1-NT peptides were synthesized and loaded in MCF-7 cells to block the interaction (TAT, a cell-penetrating peptide). After Orai1 peptide incubated with MCF-7 cells expressing GFP-NCL, the co-localization of Orai1 peptide and NCL was observed in the cell membrane and nucleus by confocal microscopy (Fig. 6A), which indicates that Orai1 peptide may play the role of disrupting the interaction between NCL and Orai1 by competitively binding NCL in the different cellular compartments. To test this possibility, we still performed the pull-down assay using biotinylated Orai1-NT peptides and cell lysate of MCF-7 overexpressing NCL-Flag with or without TAT-Orai1-NT peptides treatment. As expected, the cell-penetrating Orai1 peptide effectively reduced the co-precipitation of NCL with the biotinylated peptides (Fig. 6B, lane3). Next, we test whether TAT-Orai1 peptide (No coupling rhodamine) inhibited the NCL-meditated SOCE. MCF-7 cells transfected with GFP-NCL exhibited elevated Ca2+ influx after the addition of TG and 2 mM Ca2+ than cells transfected with GFP, but when treated with Orai1 peptides, the SOCE of GFP-NCL transfected cells was dramatically reduced (Fig. 6C, D). We also investigated whether Orai1 peptides performed the inhibitory effect on the proliferation of breast cancer cells. MTT assay revealed that Orai1 peptides treatment reversed the increased cell proliferation of breast cancer cells induced by NCL overexpression (Fig. 6E). Similarly, the EdU assay results revealed that the percentage of EdU-labeled cells was declined after Orai1 peptides treatment, suggesting peptides abolished the promoting effects of NCL on cell proliferation (Fig. 6F, G). To confirm the hypothesis that NCL promoted tumor growth by increasing calcium influx via interaction with Orai1, we subcutaneously implanted MCF-7 cells stably expression of NCL and negative control cells into nude mice to establish the subcutaneous breast cancer xenograft. NCL group was treated with 2-APB to inhibit the calcium influx or treated with Orai1 peptides to block the NCL-Orai1 interaction. As expected, NCL overexpression markedly promoted the growth of tumor volume and increased tumor weight than the negative control group. Still, both 2-APB treated group and Orai1 peptides treated group displayed a significantly smaller tumor volume compared to the NCL group (Fig. 6H–J). Taken together, these results suggested that NCL-Orai1 interaction-meditated calcium entry was crucial for breast cancer development, and blocking the NCL-Orai1 pathway may be an effective strategy for the treatment of some breast cancer subtypes.

A Co-localization of GFP-NCL (green) with rhodamine-labeled TAT-Orai1-NT peptide (red). MCF-7 cells transfected with GFP-NCL were incubated with Orai1 peptide for 1 h, then the cellular co-localization of TAT-Orai1-NT peptide and NCL was detected with confocal microscopy. B Biotin-based peptide pulldown assay to determine peptide-blocking effect of interaction between NCL and Orai1. MCF-7 cells transfected with NCL-Flag plasmid were incubated with the cell-penetrating TAT-Orai1-NT peptides. The cell lysate was bound with biotinylated Orai1-NT peptides immobilized on streptavidin beads and precipitated NCL proteins were detected by western blotting using anti-Flag antibody. C, D Measurement intracellular calcium influx using confocal microscopy in MCF-7 cells transfected with GFP-NCL and treated with Orai1-NT peptide. E MTT assay to assess the effect of Orai1-NT peptide on the proliferation of MCF-7 cells stable expression of NCL. F, G EdU staining for evaluation of the influences of Orai1 peptide on the proliferation of breast cancer cells stable expression of NCL. H Subcutaneous xenograft tumor growth in nude mice was measured. MCF-7 cells stable expression of NCL and negative control cells were transplanted to BALB/c nude mice. Mice were treated with Orai1 peptides or 2-APB. Tumor volume and weight were measured (I, J). All data are shown as mean ± SD. of three replicates. *P < 0.05. **P < 0.01. ***P < 0.001.
Discussion
Calcium signaling is an essential mediator of many processes in breast cancer progression. Calcium channels have been considered as potential drug targets in breast cancer. Orai1 has been reported to control tumorigenesis and tumor progression by virtue of the abnormal expression and activation, resulting in the imbalance of intracellular calcium homeostasis [38, 39]. Orai1 is mainly activated by calcium sensor STIM1 when calcium stores are depleted, whereas several interacting partners of Orai1 have been identified and involved in the regulation of calcium signal, suggesting that the activation mechanism and function of Orai1 still need to be clarified. In this research, we found a novel interaction between Orai1 and NCL (Figs. 1, 3).
NCL, an important nucleolar phosphoprotein, is involved in ribosomal biogenesis, rRNA processing and mRNA stability. NCL is mainly distributed in the nucleolus, but also in the nucleoplasm, cytoplasm and cell surface. Overexpression of NCL has been found in a variety of tumors, and each cell compartment of NCL could exert distinct functions in cancer progression. Our findings uncover a critical function of NCL in the development of breast cancer (Fig. 2). Recently, cell surface NCL as a receptor has attracted great attention as a potential target for cancer therapy [40]. AS1411, a DNA aptamer targeting cell surface NCL, has shown potent anti-proliferative activity in various cancer cells and entered phase II clinical trials in multiple advanced cancers [41, 42]. Moreover, various molecules have been identified as cell surface NCL-ligands involved in tumor development [43]. Our studies provided new insights into the role of cell surface NCL in breast cancer. Both the N-terminal and C-terminal of NCL interact with Orai1, which may facilitate the cell surface accumulation of NCL. Overexpression of NCL-CT exhibits a significant increase of SOCE, but the N-terminal of NCL doesn’t affect the TG-evoked SOCE (Fig. 4). NCL has been reported to be a calcium-binding protein, and the highly acidic N-terminal contributes to binding Ca2+. So, the N-terminal of NCL may play a function similar to STIM1, sensing the calcium concentration change in the nucleus and assisting the interaction between NCL-CT and Orai1. We also found that knockdown of NCL expression caused the decrease of calcium in the cell nucleus after TG treatment (Fig. S6). So far, the regulatory mechanism of nuclear calcium is still very unclear. We speculate that NCL may play a role in regulating nuclear calcium. When the ER calcium store is depleted and nuclear calcium is reduced, NCL may sense the change of calcium concentration via its N-terminal, resulting in a conformation change and translocation of NCL from the nucleus to the cytoplasm and cell surface where it interacts with Orai1 to meditate calcium influx replenishing the calcium store and nuclear calcium. However, its underlying molecular mechanisms need to be further clarified.
Accumulating evidence of NCL in cancer development and cancer therapy suggests that NCL is a candidate biomarker and a promising target for cancer treatment. Our studies have demonstrated that NCL plays a crucial role in the proliferation of breast cancer cells by regulating the SOCE. Orai1 inhibitor 2-APB can significantly inhibit NCL-meditated calcium influx and abolish the promoting effects of NCL on breast cancer cells proliferation (Fig. 5). More importantly, the synthesized Orai1 peptide specifically targeting NCL and blocking NCL-Orai1 interaction, attenuates the growth of breast cancer cells through the reduction of NCL-meditated SOCE. Move over, peptides exhibit the strong anti-tumor activity in a xenograft mouse model (Fig. 6). Thus, the NCL-Orai1 interaction may be a valuable target for controlling calcium homeostasis and the development of breast cancer.
Conclusions
In summary, we identified NCL as a key regulator for calcium homeostasis in breast cancer cells via activation of Orai1. Blocking the Orai1 channel or NCL-Orai1 interaction inhibits the promoting effects of NCL on breast cancer cells proliferation (Fig. 7). Our results provide the novel potential therapeutic targets for breast cancer treatment.

A NCL proteins shuttle from nucleolus to cytoplasm and cell membrane, and then interact with calcium channel Orai1 at the plasma membrane via the C-terminal domain, which results in activation of Orai1 channel and calcium entry, eventually leading to activation of calcium related signaling pathways and promotion breast cancer cell proliferation. B Orai1 inhibitor 2-APB and synthesized Orai1-NT peptides blocking NCL/Orai1 interaction could inhibit NCL-meditated calcium influx, partially restore intracellular calcium homeostasis and inhibit the proliferation of breast cancer cells.
Data availability
The data and materials of this study are available from the corresponding author upon reasonable request.
References
Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Prim. 2019;5:66.
Pratt SJP, Hernández-Ochoa E, Martin SS. Calcium signaling: breast cancer’s approach to manipulation of cellular circuitry. Biophys Rev. 2020;12:1343–59.
So CL, Saunus JM, Roberts-Thomson SJ, Monteith GR. Calcium signalling and breast cancer. Semin Cell Dev Biol. 2019;94:74–83.
Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol. 2014;171:945–60.
Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium-cancer signalling nexus. Nat Rev Cancer 2017;17:367–80.
Marchi S, Pinton P. Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol. 2016;29:1–6.
Putney JW. The physiological function of store-operated calcium entry. Neurochem Res. 2011;36:1157–65.
Tojyo Y, Morita T, Nezu A, Tanimura A. Key components of store-operated Ca2+ entry in non-excitable cells. J Pharmacol Sci. 2014;125:340–6.
Jardin I, Lopez JJ, Salido GM, Rosado JA. Store-operated Ca(2+) entry in breast cancer cells: remodeling and functional role. Int J Mol Sci. 2018;19:4053.
Hodeify R, Yu F, Courjaret R, Nader N, Dib M, Sun L et al. Regulation and Role of Store-operated Ca(2+) Entry in Cellular Proliferation. In: Kozak JA, Putney JW, Jr. (eds). Calcium Entry Channels in Non-Excitable Cells. CRC Press/Taylor & Francis © 2017 by Taylor & Francis Group, LLC.: Boca Raton (FL), 2018, pp 215–40.
Hammad AS, Machaca K. Store operated calcium entry in cell migration and cancer metastasis. Cells 2021;10:1246.
Yeung PS, Yamashita M, Prakriya M. Molecular basis of allosteric Orai1 channel activation by STIM1. The. J Physiol. 2020;598:1707–23.
Lunz V, Romanin C, Frischauf I. STIM1 activation of Orai1. Cell Calcium. 2019;77:29–38.
Hogan PG. The STIM1-ORAI1 microdomain. Cell Calcium. 2015;58:357–67.
**e J, Pan H, Yao J, Zhou Y, Han W. SOCE and cancer: Recent progress and new perspectives. Int J Cancer. 2016;138:2067–77.
Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–34.
Wang JY, Sun J, Huang MY, Wang YS, Hou MF, Sun Y, et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene 2015;34:4358–67.
Kim JH, Lkhagvadorj S, Lee MR, Hwang KH, Chung HC, Jung JH, et al. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochemical biophysical Res Commun. 2014;448:76–82.
Umemura M, Bal**nyam E, Feske S, De Lorenzo MS, **e LH, Feng X, et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PloS One. 2014;9:e89292.
Kondratska K, Kondratskyi A, Yassine M, Lemonnier L, Lepage G, Morabito A, et al. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochimica et Biophysica Acta. 2014;1843:2263–9.
Srikanth S, Gwack Y. Orai1, STIM1, and their associating partners. J Physiol. 2012;590:4169–77.
Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010;143:84–98.
Srikanth S, Jung HJ, Kim KD, Souda P, Whitelegge J, Gwack Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol. 2010;12:436–46.
Albarran L, Lopez JJ, Amor NB, Martin-Cano FE, Berna-Erro A, Smani T, et al. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci Rep. 2016;6:24452.
Tajrishi MM, Tuteja R, Tuteja N. Nucleolin: The most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol. 2011;4:267–75.
Abdelmohsen K, Gorospe M. RNA-binding protein nucleolin in disease. RNA Biol. 2012;9:799–808.
Chen Z, Xu X. Roles of nucleolin. Focus on cancer and anti-cancer therapy. Saudi Med J. 2016;37:1312–8.
Jia W, Yao Z, Zhao J, Guan Q, Gao L. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci. 2017;186:1–10.
Berger CM, Gaume X, Bouvet P. The roles of nucleolin subcellular localization in cancer. Biochimie 2015;113:78–85.
Tsou JH, Chang KY, Wang WC, Tseng JT, Su WC, Hung LY, et al. Nucleolin regulates c-Jun/Sp1-dependent transcriptional activation of cPLA2alpha in phorbol ester-treated non-small cell lung cancer A549 cells. Nucleic acids Res. 2008;36:217–27.
Zhang J, Tsaprailis G, Bowden GT. Nucleolin stabilizes Bcl-X L messenger RNA in response to UVA irradiation. Cancer Res. 2008;68:1046–54.
Ginisty H, Sicard H, Roger B, Bouvet P. Structure and functions of nucleolin. J Cell Sci. 1999;112:761–72.
Gilchrist JS, Abrenica B, DiMario PJ, Czubryt MP, Pierce GN. Nucleolin is a calcium-binding protein. J Cell Biochem. 2002;85:268–78.
Losfeld ME, Khoury DE, Mariot P, Carpentier M, Krust B, Briand JP, et al. The cell surface expressed nucleolin is a glycoprotein that triggers calcium entry into mammalian cells. Exp Cell Res. 2009;315:357–69.
Hovanessian AG, Soundaramourty C, El Khoury D, Nondier I, Svab J, Krust B. Surface expressed nucleolin is constantly induced in tumor cells to mediate calcium-dependent ligand internalization. PloS One. 2010;5:e15787.
Ke J, Gu C, Zhang H, Liu Y, Zhang W, Rao H, et al. Nucleolin Promotes Cisplatin Resistance in Cervical Cancer by the YB1-MDR1 Pathway. J Oncol. 2021;2021:9992218.
Niu L, Wu F, Li K, Li J, Zhang SL, Hu J et al. STIM1 interacts with termini of Orai channels in a sequential manner. J cell Sci. 2020;133:jcs239491.
Bhargava A, Saha S. T-Type voltage gated calcium channels: a target in breast cancer? Breast Cancer Res Treat. 2019;173:11–21.
Chang Y, Roy S, Pan Z. Store-operated calcium channels as drug target in gastroesophageal cancers. Front Pharmacol. 2021;12:668730.
Koutsioumpa M, Papadimitriou E. Cell surface nucleolin as a target for anti-cancer therapies. Recent Pat Anti-cancer Drug Discov. 2014;9:137–52.
Soundararajan S, Chen W, Spicer EK, Courtenay-Luck N, Fernandes DJ. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008;68:2358–65.
Rosenberg JE, Bambury RM, Van Allen EM, Drabkin HA, Lara PN Jr., Harzstark AL, et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investig N. Drugs. 2014;32:178–87.
Fujiki H, Watanabe T, Suganuma M. Cell-surface nucleolin acts as a central mediator for carcinogenic, anti-carcinogenic, and disease-related ligands. J Cancer Res Clin Oncol. 2014;140:689–99.
Acknowledgements
We thank Dr. Yimeng Zhu for valuable discussions and revised English grammar. This work was supported by the National Natural Science Foundation of China (Grant no. 81702639), The Natural Science Foundation of Hubei Provincial Department of Education (Grant no. D20212102), Free Exploration Project of Hubei University of Medicine (Grant no. FDFR201901), Innovative Research Program for Graduates of Hubei University of Medicine (Grant nos. YC2020007, YC2021009).
Author information
Authors and Affiliations
Contributions
FYW and SL designed and supervised the research. CMG, WHZ and EZY performed the experiments and analyzed the data. JK and ZYZ performed the animal experiments. CYG performed the immunohistochemistry experiments. XW and SYW gave technical support. FYW wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The study protocol was approved by the ethics committee of Hubei University of Medicine (No. 2021TH063). The animal study was approved by the Hubei University of Medicine Animal Ethics Committee (No. 201716).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gu, C., Zhang, W., Yang, E. et al. Blockage of Orai1-Nucleolin interaction meditated calcium influx attenuates breast cancer cells growth. Oncogenesis 11, 55 (2022). https://doi.org/10.1038/s41389-022-00429-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-022-00429-z
- Springer Nature Limited