
Abstract
Increasing evidence indicates that DNA damage-induced apoptosis suppressor (DDIAS) is an oncogenic protein that is highly expressed in a variety of cancers, including colorectal cancer, lung cancer, breast cancer, and hepatocellular carcinoma (HCC). The discovery of DDIAS as a novel therapeutic target and its role in human cancer biology is fascinating and noteworthy. Recent studies have shown that DDIAS is involved in tumorigenesis, metastasis, DNA repair and synthesis, and drug resistance and that it plays multiple roles with distinct binding partners in several human cancers. This review focuses on the function of DDIAS and its regulatory proteins in human cancer as potential targets for cancer therapy, as well as the development and future prospects of DDIAS inhibitors.
Similar content being viewed by others

Introduction
DNA damage-induced apoptosis suppressor (DDIAS) was first discovered as a human homolog (hNoxin) of mouse noxin via genomic analysis of colorectal cancer patients and large-scale siRNA screening aimed at searching for cancer-related genes1,2. DDIAS was named based on its antiapoptotic properties in response to DNA repair in cancer cells.
DDIAS is highly expressed in several human cancers, including colorectal cancer, lung cancer, breast cancer and hepatocellular carcinoma (HCC), and stimulates cancer cell proliferation and cell cycle progression2,3,4,5. DDIAS plays a vital role in tumorigenesis, metastasis, DNA repair and drug resistance by inhibiting cell death mediated by DNA damage agents, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and gefitinib in lung cancer2,5,6,7,8. In HCC, DNA copy number amplification of DDIAS has been observed3. Interestingly, DDIAS interacts with various binding partners to drive numerous processes in the membrane, cytoplasm and nucleus through various extracellular signals.
Although DDIAS studies on the novel cancer-related processes are still lacking, DDIAS appears to play a key role in carcinogenesis, particularly in lung, liver and colorectal cancers. DDIAS is not a well-known gene to most cancer researchers even though it is noteworthy as a novel cancer therapeutic target. In this review, we discuss various aspects of DDIAS, including transcriptional regulation, degradation, DNA repair, and resistance to apoptosis, and suggest prospective cancer treatments by inhibiting DDIAS-related cellular functions.
Molecular features of DDIAS
The DDIAS gene (noxin, C11orf82, GeneID220042) encodes 998 amino acids and consists of six exons with a translation initiation site at the third exon. DDIAS features a DNA-binding domain C (DBD C) in the N-terminal region (amino acids 8–123), which is also found in replication protein A (RPA), a nuclear single-stranded DNA-binding protein. DBD C is essential for forming heterotrimeric complexes with RPA1 with RPA2 and RPA3 for replication, recombination and repair, and interactions with nuclear proteins9,10,11. The DDIAS gene is located on human chromosome 11 and is conserved (70% similarity at the DNA level) in the rat and mouse genomes (rat chromosome 1 and mouse chromosome 7). Although DDIAS has been recognized as a human homolog of mouse nitric oxide-inducible (noxin), a sequence comparison of the DDIAS protein with mouse noxin reveals high homology only in the N-terminal region containing the DBD C (80.5%; amino acids 1–123) and lower homology throughout the remainder of the protein sequences (33.2%; amino acids 124–998), despite conserved several sequences (Fig. 1). The DDIAS protein sequence is enriched with serine (132 serine residues), indicating that phosphorylation of these residues might be a potential modification. Similar to mouse noxin, DDIAS carries putative sites of phosphorylation by DNA-PK, ATM, cdc2, CDK5, CKII, p38 mitogen activated protein kinase (p38MAPK), RSK, PKA, and PKC (NetPhos 3.1). Recently, Akimov and colleagues reported that DDIAS is ubiquitinated at lysine386, lysine500 and lysine807, as previously discovered using the UbiSite method12 (Fig. 1).

a, b Comparison of rat, mouse, and human DDIAS proteins. Sequence comparison of human DDIAS protein with mouse DDIAS (noxin) revealed high homology in the N-terminal region containing the DBD C (80.5%; amino acids 1–123) and less homology throughout the rest of the proteins (33.2%; amino acids 124–998). Nuc, nuclear localization signals (red); Zn, zinc-finger-like domain (pink); DNA-PK, DNA-PK phosphorylation consensus sequences (yellow); Akt or ATM, Akt or ATM phosphorylation consensus sequences (yellow); DBD C, DNA binding domain C (Pink); MED29, mediator complex subunit 29 (blue). Potential ubiquitination sites in DDIAS (K386, K500, and K807) are shown.
Regulation of DDIAS expression
DDIAS expression patterns
In normal human tissues, low expression of DDIAS has been detected in the lung, stomach, thymus, colon and heart, while high expression has been detected in the human testis, pancreas and prostate2. However, DDIAS mRNA levels are significantly higher in lung, breast, and colorectal cancer tissues and cancer cell lines than in normal tissues or cells2,3. HCC is characterized by DDIAS overexpression with DNA copy number amplification on chromosome 11q14.15. DDIAS expression is induced by ultraviolet (UV) irradiation, and its level is highest in the S phase of the cell cycle in cancer and normal cells. Similar to DDIAS, mouse noxin is highly expressed in the testis1,2. Similarly, numerous stressors, such as gamma ray irradiation, UV irradiation, NO donors, hydrogen peroxide, adriamycin, and cytokines, stimulate the production of mouse noxin. However, mouse noxin expression is highest in cells in the G2/M phase or after exposure to nocodazole, a G2/M arrest inducer.
The Human Protein Atlas revealed that DDIAS is found mainly in the cytoplasm. Endogenous DDIAS is present in the cytoplasm of lung cancer cells and tissues, as well as the nucleus of HCC cells4,5,13. In NIH3T3 cells, endogenous mouse noxin is found in the cytoplasm and the nucleus; however, it accumulates in the nucleus in response to exposure to the nitric oxide donor SNAP1.
Transcriptional regulation of DDIAS
DDIAS is a target gene of nuclear factor of activated T cells 1 (NFATc1, NFAT2) in lung cancer6. In a DDIAS promoter analysis, potential binding sites for various transcription factors, such as p300, SP1, C/EBP, or NFAT, were identified. Among these transcription factors, dephosphorylated NFATc1 activates the transcription of DDIAS by binding to NFAT consensus sequences in the DDIAS promoter. DDIAS gene expression is stimulated by phorbol 12-myristate 13-acetate and the calcium ionophore A23187 and suppressed by the calcineurin inhibitor cyclosporin A, which activate and inhibit NFATc1, respectively (Fig. 2a). Additionally, tissue array immunostaining revealed a correlation between DDIAS and NFATc1 expression in human lung cancers6. Despite the importance of the NFAT family in the immune response, recent studies have indicated that activation or overexpression of NFATc1 in human solid tumors and hematological malignancies is associated with tumor progression14,15.

a Activated NFAT2 regulates DDIAS transcription. b In response to EGF, MEK5/ERK5/MEF2B pathways induce DDIAS expression. c DDIAS ubiquitination mediated by HSP70/CHIP regulates DDIAS protein turnover.
Although DDIAS expression is induced by UV irradiation of normal and cancer cells2, the mechanism underlying DDIAS-mediated transcriptional regulation by UV irradiation is not fully understood. UV irradiation generally stimulates mitogen-activated protein kinase (MAPK) and ATM pathways and the activation of transcription factors such as p53, NF-kB, AP-1, NFAT, and Nrf216,17. Similarly, the induction of mouse noxin function mediated by stress stimuli, including UV irradiation, depends on p531.
In addition to UV exposure, DDIAS expression is induced by serum or epidermal growth factor (EGF)2,13. Previous studies have demonstrated that extracellular signal-regulated kinase 5 (ERK5) phosphorylates myocyte enhancer factor-2 (MEF2) family proteins and serum glucocorticoid-inducible kinase, all of which are essential for entry into the S phase of the cell cycle18,19. DDIAS expression is induced by ERK5 and MEF2 in response to EGF13,20. Genetic or pharmacological inhibition of ERK5 suppresses DDIAS expression by EGF exposure. The overexpression of constitutively active MEK5 enhances DDIAS expression (Fig. 2b). In chromatin immunoprecipitation (ChIP) assays, MEF2B (a downstream target of ERK5) exhibited sequence-specific binding to the MEF2-binding site in the DDIAS promoter after EGF treatment. Moreover, overexpression of MEF2B increased the EGF-mediated induction of DDIAS expression, whereas knockdown of MEF2B attenuated this effect.
Posttranslational regulation of DDIAS
DDIAS stability is regulated by E3 U-box-dependent ubiquitin ligase carboxyl terminus of HSP70-interacting protein (CHIP)-mediated proteasomal degradation21. We first identified CHIP as an interacting partner of DDIAS by yeast two-hybrid screening. E3 ubiquitin ligase CHIP physically associates with both the N- and C-terminal regions of DDIAS, allowing this protein to be targeted for proteasomal degradation and thereby reducing the DDIAS half-life in the cytoplasm (Fig. 2c). CHIP deletion study demonstrated a tetratricopeptide repeat (TPR) domain and the U-box are essential for DDIAS ubiquitination. HSP70-bound DDIAS is recruited to the CHIP E3 ligase via the TPR domain, suggesting that DDIAS is a client protein of HSP70. Since CHIP is a chaperone-associated U box-containing E3 ligase, it depends on Hsp70/Hsp90 chaperones. These chaperones interact with oncogenic clients such as c-Myc, hypoxia inducible factor 1a (HIF-1a), NF-kB/p65, and DDIAS21,22,23,24, implying that CHIP is a tumor suppressor.
Molecular mechanisms of DDIAS in Cancers
DDIAS executes a variety of cellular tasks with its different binding partners (Table 1). High expression of DDIAS in cancer contributes to malignancies mediated via a variety of mechanisms (Fig. 3). The functions of DDIAS associated with cancer are discussed herein.

a DDIAS promotes DNA synthesis and DSB (double-strand break) repair in human cancers. b DDIAS enhances the proliferation and metastasis of cancer cells mediated by STAT3 phosphorylation by competing with PTPRM. c DDIAS prevents cancer cells from undergoing apoptosis by inhibiting the p38 MAPK/p53/p21 pathway and TRAIL-mediated DISC formation. d DDIAS contributes to TRAIL and tamoxifen resistance.
DNA synthesis and repair
DDIAS overexpression accelerates the G1-S phase transition by enhancing DNA synthesis in HCC5. DDIAS interacts with DNA polymerase α, suggesting that DDIAS may boost de novo DNA synthesis by promoting the formation of DNA polymerase-primase complexes (Fig. 3a). DDIAS overexpression promotes cellular proliferation, colony formation, cellular migration and in vivo tumorigenicity, whereas DDIAS knockdown attenuates these effects.
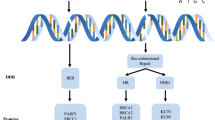
On the basis of computational approaches such as evolutionary rate covariation, a recent study revealed 17,487 mammalian genes coevolved in six distinct DNA repair pathways. Among these coevolved proteins, DDIAS was identified as a novel factor in double-strand break (DSB) repair based on its coevolution with homologous recombination (HR)25. It is involved in a DSB repair mechanism mediating nonhomologous end-joining26,27. DDIAS depletion resulted in DSB accumulation, as indicated by ATM kinase activation and 53BP1 foci induction, and defective HR (Fig. 3a). Similarly, a previous report showed an increase in H2AXγ, a marker of DNA DSBs, and comet formation, a measure of DNA strand breaks, with cells depleted of DDIAS2. Furthermore, DDIAS carries an oligonucleotide/oligosaccharide-binding-fold domain similar to the single-strand DNA-binding domain of RPA2. This domain is required for replication, recombination and repair processes such as HR28, providing evidence supporting the involvement of DDIAS in DNA repair.
Proliferation and metastasis of cancer cells
The activation of signal transducer and activator of transcription 3 (STAT3) plays a critical role in cancer cell proliferation, survival, metastasis, and self-renewal29,30,31,32. Phosphorylated STAT3 enters the nucleus and induces the transcription of target genes, including survivin, Bcl-2, Mcl-1, c-Myc, cyclin D1, slug, and matrix metalloproteinase-233, Nakaya, N. et al. noxin, a novel stress-induced gene involved in cell cycle and apoptosis. Mol. Cell. Biol. 27, 5430–5444 (2007). Won, K. J. et al. Human Noxin is an anti-apoptotic protein in response to DNA damage of A549 non-small cell lung carcinoma. Int. J. Cancer 134, 2595–2604 (2014). Zhang, X. et al. Noxin promotes proliferation of breast cancer cells via P38-ATF2 signaling pathway. Tumour Biol. 39, 1010428317705515 (2017). Liu, N. et al. DDIAS promotes invasion and proliferation of non-small cell lung cancer and predicts poor survival of lung cancer patients. Int. J. Clin. Exp. Pathol. 10, 11506–11515 (2017). Zhang, Z. Z., Huang, J., Wang, Y. P., Cai, B. & Han, Z. G. NOXIN as a cofactor of DNA polymerase-primase complex could promote hepatocellular carcinoma. Int. J. Cancer 137, 765–775 (2015). Im, J. Y. et al. DNA damage-induced apoptosis suppressor (DDIAS), a novel target of NFATc1, is associated with cisplatin resistance in lung cancer. Biochim. Biophys. Acta 1863, 40–49 (2016). Im, J. Y. et al. DDIAS suppresses TRAIL-mediated apoptosis by inhibiting DISC formation and destabilizing caspase-8 in cancer cells. Oncogene 37, 1251–1262 (2018). Im, J. Y. et al. DDIAS promotes STAT3 activation by preventing STAT3 recruitment to PTPRM in lung cancer cells. Oncogenesis 9, 1 (2020). Dueva, R. & Iliakis, G. Replication protein A: a multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2, zcaa022 (2020). Wold, M. S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92 (1997). Iftode, C., Daniely, Y. & Borowiec, J. A. Replication protein A (RPA): the eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 34, 141–180 (1999). Akimov, V. et al. UbiSite approach for comprehensive map** of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 25, 631–640 (2018). Im, J. Y. et al. DNA damage induced apoptosis suppressor (DDIAS) is upregulated via ERK5/MEF2B signaling and promotes beta-catenin-mediated invasion. Biochim. Biophys. Acta 1859, 1449–1458 (2016). Mancini, M. & Toker, A. NFAT proteins: emerging roles in cancer progression. Nat. Rev. Cancer 9, 810–820 (2009). Muller, M. R. & Rao, A. NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 10, 645–656 (2010). Lakin, N. D. & Jackson, S. P. Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655 (1999). Lopez-Camarillo, C. et al. Protein kinases and transcription factors activation in response to UV-radiation of skin: implications for carcinogenesis. Int. J. Mol. Sci. 13, 142–172 (2012). Kato, Y. et al. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 16, 7054–7066 (1997). Kato, Y. et al. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 395, 713–716 (1998). Im, J. Y. et al. Data on the transcriptional regulation of DNA damage induced apoptosis suppressor (DDIAS) by ERK5/MEF2B pathway in lung cancer cells. Data Brief. 9, 257–261 (2016). Won, K. J. et al. Stability of the cancer target DDIAS is regulated by the CHIP/HSP70 pathway in lung cancer cells. Cell Death Dis. 8, e2554 (2017). Paul, I., Ahmed, S. F., Bhowmik, A., Deb, S. & Ghosh, M. K. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene 32, 1284–1295 (2013). Ferreira, J. V. et al. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 9, 1349–1366 (2013). Wang, S. et al. CHIP functions as a novel suppressor of tumour angiogenesis with prognostic significance in human gastric cancer. Gut 62, 496–508 (2013). Brunette, G. J., Jamalruddin, M. A., Baldock, R. A., Clark, N. L. & Bernstein, K. A. Evolution-based screening enables genome-wide prioritization and discovery of DNA repair genes. Proc. Natl Acad. Sci. U.S.A. 116, 19593–19599 (2019). Scully, R., Panday, A., Elango, R. & Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714 (2019). Chapman, J. R., Taylor, M. R. & Boulton, S. J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510 (2012). Chen, R. & Wold, M. S. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 36, 1156–1161 (2014). Bromberg, J. F. et al. Stat3 as an oncogene. Cell 98, 295–303 (1999). Yu, H. & Jove, R. The STATs of cancer-new molecular targets come of age. Nat. Rev. Cancer 4, 97–105 (2004). Yu, H., Pardoll, D. & Jove, R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 (2009). Huynh, J., Chand, A., Gough, D. & Ernst, M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat. Rev. Cancer 19, 82–96 (2019). Bhattacharya, S., Ray, R. M. & Johnson, L. R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 392, 335–344 (2005). **e, T. X. et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 23, 3550–3560 (2004). Gritsko, T. et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 12, 11–19 (2006). Zhong, Z., Wen, Z. & Darnell, J. E. Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98 (1994). Hillmer, E. J., Zhang, H., Li, H. S. & Watowich, S. S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 31, 1–15 (2016). Shuai, K. & Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3, 900–911 (2003). Takeiwa, T. et al. PSPC1 is a potential prognostic marker for hormone-dependent breast cancer patients and modulates RNA processing of ESR1 and SCFD2. Sci. Rep. 12, 9495 (2022). Mitobe, Y. et al. PSF promotes ER-positive breast cancer progression via posttranscriptional regulation of ESR1 and SCFD2. Cancer Res. 80, 2230–2242 (2020). Yeh, H. W. et al. PSPC1 mediates TGF-beta1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat. Cell Biol. 20, 479–491 (2018). Lang, Y. D. et al. PSPC1-interchanged interactions with PTK6 and beta-catenin synergize oncogenic subcellular translocations and tumor progression. Nat. Commun. 10, 5716 (2019). Zhang, L. & Fang, B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 12, 228–237 (2005). Trivedi, R. & Mishra, D. P. Trailing TRAIL resistance: novel targets for TRAIL sensitization in cancer cells. Front. Oncol. 5, 69 (2015). Roberts, T. C., Langer, R. & Wood, M. J. A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 19, 673–694 (2020). Hu, B. et al. Therapeutic siRNA: state of the art. Signal Transduct. Target. Ther. 5, 101 (2020). Muralidharan, R. et al. HuR-targeted nanotherapy in combination with AMD3100 suppresses CXCR4 expression, cell growth, migration and invasion in lung cancer. Cancer Gene Ther. 22, 581–590 (2015). Muralidharan, R. et al. Tumor-targeted nanoparticle delivery of HuR siRNA inhibits lung tumor growth in vitro and in vivo by disrupting the oncogenic activity of the RNA-binding protein HuR. Mol. Cancer Ther. 16, 1470–1486 (2017). Im, J. Y. et al. DGG-100629 inhibits lung cancer growth by suppressing the NFATc1/DDIAS/STAT3 pathway. Exp. Mol. Med. 53, 643–653 (2021). Hong, D. et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 7, 314ra185 (2015). Fagard, R., Metelev, V., Souissi, I. & Baran-Marszak, F. STAT3 inhibitors for cancer therapy: Have all roads been explored? JAKSTAT 2, e22882 (2013). Dong, J., Cheng, X. D., Zhang, W. D. & Qin, J. J. Recent update on development of small-molecule STAT3 inhibitors for cancer therapy: from phosphorylation inhibition to protein degradation. J. Med. Chem. 64, 8884–8915 (2021). Yoon, S. H., Kim, B. K., Kang, M. J., Im, J. Y. & Won, M. Miconazole inhibits signal transducer and activator of transcription 3 signaling by preventing its interaction with DNA damage-induced apoptosis suppressor. Cancer Sci. 111, 2499–2507 (2020). Ershov, N. I. et al. Consequences of early life stress on genomic landscape of H3K4me3 in prefrontal cortex of adult mice. BMC Genom. 19, 93 (2018). Chiarelli, N., Carini, G., Zoppi, N., Ritelli, M. & Colombi, M. Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients’ skin fibroblasts. PLoS One 14, e0211647 (2019). Charkoftaki, G. et al. Identification of dose-dependent DNA damage and repair responses from subchronic exposure to 1,4-dioxane in mice using a systems analysis approach. Toxicol. Sci. 183, 338–351 (2021).References
Acknowledgements
This work was supported by the National Research Foundation (NRF-2017R1A2B2011936) and the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5192221).
Author information
Authors and Affiliations
Contributions
J.Y.I., M.J.K., and B.K.K. collected the literature and conceived the review. J.Y.I. wrote the manuscript. J.Y.I. and M.W. revised and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Im, JY., Kang, MJ., Kim, BK. et al. DDIAS, DNA damage-induced apoptosis suppressor, is a potential therapeutic target in cancer. Exp Mol Med 55, 879–885 (2023). https://doi.org/10.1038/s12276-023-00974-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00974-6
- Springer Nature Limited