

Abstract
Silicate glasses are durable materials, but are they sufficiently durable to confine highly radioactive wastes for hundreds of thousands years? Addressing this question requires a thorough understanding of the mechanisms underpinning aqueous corrosion of these materials. Here we show that in silica-saturated solution, a model glass of nuclear interest corrodes but at a rate that dramatically drops as a passivating layer forms. Water ingress into the glass, leading to the congruent release of mobile elements (B, Na and Ca), is followed by in situ repolymerization of the silicate network. This material is at equilibrium with pore and bulk solutions, and acts as a molecular sieve with a cutoff below 1 nm. The low corrosion rate resulting from the formation of this stable passivating layer enables the objective of durability to be met, while progress in the fundamental understanding of corrosion unlocks the potential for optimizing the design of nuclear glass-geological disposal.
Similar content being viewed by others
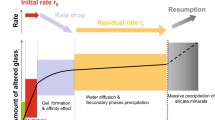


Introduction
The behaviour of silicate glasses over geological timescales raises challenging scientific issues. One of the most prominent topics with a high social impact is the safe management of highly radioactive wastes originating from spent nuclear fuel reprocessing that, among others, depends on our capacity to predict the long-term corrosion rate of borosilicate glass1,2,3. Geochemists face similar fundamental problems with basaltic glass when calculating the chemical mass balance of the oceans4 or when attempting to assess potential CO2 sequestration by silicate rocks5. Current rate laws still cannot accurately predict the long-term behaviour of synthetic or natural silicate glasses over geological timescales mostly because of the existence of surface layers between pristine glass and bulk solution that both affect the transport of reactive species and change fluid properties at the glass surface compared with that of the bulk6,7. Surface layers are made of amorphous and crystalline metastable phases, formed by in situ condensation and precipitation reactions from aqueous species8. Glass dissolves following the Ostwald rule of stages, forming a series of alteration products progressively evolving towards thermodynamically stable phases6,9. Depending on the glass composition and on the conditions (for example, temperature, pH, solution composition and flow rate), this transformation into stable compounds can vary from hours to millions of years6,10,11!
In this study, glass corrosion mechanisms were investigated from macroscopic to atomistic scale, through isotopically tagged corrosion experiments performed with international simple glass (ISG)—a six-oxide borosilicate glass used as a reference material by the nuclear glass community1—and the use of complementary analytical techniques to provide evidence of the long-term rate-limiting mechanisms. The main experiment was performed with 16 coupons of ISG glass (with isotopes at the natural abundance) altered for 1 year in a static mode at 90 °C, pH90 °C 7, in a solution initially saturated with amorphous 29SiO2 (Supplementary Fig. 1). Previous experiments showed that when altered in deionized water the leaching solution of ISG glass eventually reaches the equilibrium with amorphous silica (SiO2am) but this requires several years12. This starting solution thus enables the first transient stages of glass corrosion to be bypassed and focus placed on the processes governing the long-term rate. In parallel, a similar experiment was conducted with glass powder until its complete alteration, in order to examine the structural changes within the silicate network. The choice of pH 7 was motivated by two reasons. First, after similar investigations carried out at pH 9 and 11.5, it was found that results at pH 7 were the most convincing because glass corrodes about four times faster at pH 7 than at pH 9, although the fundamental processes are the same13. This led to larger amounts of material available for in-depth characterization. Second, a pH of 7 is typical of many natural waters; for instance, the water in equilibrium with the claystone studied in France for the storage of nuclear wastes is pH 7.3 at room temperature14. Together, these studies improve our understanding of silicate glass corrosion processes, by linking dissolution kinetics and structural modifications following water ingress into the solid. At this pH, the glass is passivated by a self-healing and poorly hydrated silica layer in which water is trapped in subnanometric pores. Conclusions can be extended at least until pH 9.5, leading to general recommendations for the safe management of highly radioactive waste glass.
Results
Glass still corrodes despite silica saturation conditions
Despite the fact that the solution was initially saturated with respect to amorphous silica, the ISG glass still corrodes. This phenomenon occurred under very stable conditions: pH was kept constant (Fig. 1a) and the Si concentration did not change for the entire duration of the test (Fig. 1b). In these conditions, the glass dissolution rate, given by the release time of B, Na or Ca (the three elements behave similarly), dramatically diminished during the experiment (Fig. 1c,d). The rate decrease is ~3.7 orders of magnitude from the first hours (~500 nm d−1, which is close to the maximum rate measured far from equilibrium15), to 363 days (around 0.1 nm d−1). In the meantime, Si, Al and Zr remained almost undissolved (at 363 days, the normalized loss of Al and Zr are 100 and 1,300 times smaller than that of B, respectively) (Supplementary Table 1). In addition, as the 29Si/28Si ratio remained stable over time, it could be concluded that 29Si(aq) did not—or if so, only weakly—interact with Si from the glass during corrosion (Fig. 1b). From this, it seems that the three mobile elements behaved independently from the three low-soluble cations constituting the glassy network. This might seem at odds with the most of accepted models that attributed the drop in the corrosion rate to an increasing activity of the dissolved silica16,17,18,19.

Time evolution of the solution pH (a), the concentration of Si, 29Si/28Si and 29Si/30Si ratios (b), the equivalent thickness of altered glass (ETh) calculated from B release into solution (c) and the glass dissolution rate (d). The pH90 °C was maintained at 7±0.25 by regular additions of diluted HNO3. Boron was used to calculate glass corrosion thickness and rate. This element is known to be a good corrosion tracer as it is very soluble and only slightly retained in the alteration layer (as verified by ToF-SIMS and EFTEM analyses).
Mobile species display local gradients
Deeper insights into the glass corrosion mechanisms were obtained from a detailed characterization of the altered glass coupons withdrawn at 7, 209 and 363 days. For the three samples, the alteration layer displays three sublayers (Fig. 2). From the solution to the pristine glass a gradient area can first be noticed, which B, Na and Ca concentrations dropped similarly. As time-of-flight secondary ion mass spectroscopy (ToF-SIMS) is known to potentially broaden chemical profiles especially in the case of rough or tilted interfaces20, B profile was also characterized by transmission electron microscope (TEM) in order to avoid misinterpretation. Energy-filtered TEM (EFTEM) B map** performed on a focused ion beam cross-section prepared with the 209-day sample confirmed the position and the width of the B profile given by ToF-SIMS (Fig. 2a). ToF-SIMS also revealed that B, Na and Ca gradients are well anticorrelated with that of H (Fig. 2b and Supplementary Fig. 2). This pattern is generally attributed to ion exchange between exogenous positively charged species (H+, H3O+ and K+) and Na+, Ca++ from the glass and accompanied by the fast hydrolysis of B–O bonds21,22. Beyond this gradient area, B and Ca concentrations slowly decreased to zero, whereas Na remained at a low but constant concentration, likely to act as a charge compensator along with K for fourfold coordinated Al species and sixfold coordinated Zr species23. This large, central area showed a nearly flat concentration of all the glass constituents. Finally, an external area is visible, mainly characterized by significant changes in Si isotopic ratios (Fig. 2c).

(a) EFTEM map** and the superimposed profile of B concentration (normalized to that of the pristine glass). (b) The H profile (not-normalized) displaying an anticorrelated steeper gradient than that of the other mobile species of the glass (B, Ca and Na). (c) 29Si/28Si ratio exhibiting a slight enrichment in the first 250 nm of the alteration layer (this depth is possibly overestimated according to the caveat given in Methods) and then a constant value a few percent above the natural abundance (1(±1)% at 7 days, 5(±1)% at 209 days and 4(±1)% at 363 days). This ratio has to be compared with that of the bulk solution (~30). (d,e) Normalized profiles of sparingly soluble glass formers (Si and Al) and soluble elements (Na, Ca and B). Si and Al remain at a near constant concentration in the alteration layer, whereas mobile species (B, Ca and Na) are leached out of the layer except near the reaction front, where a gradient is clearly visible. Note that K is highly present in the alteration layer, but it is not displayed because its signal was saturated.
The alteration layer is not a precipitate
In the central area region the 29Si/28Si ratio plateaus at a few percent above the natural abundance (Fig. 2c). This slight enrichment of Si species supplied by the solution increased and stayed constant between 209 and 363 days. At the steady state, it represents one atom of Si supplied by the solution per 600 coming from the glass. This result suggests that the central part of the alteration layer is not a diffusion barrier for Si(aq) as diffusion-limiting process would have led to chemical gradients. Therefore, one can assume that a thermodynamic equilibrium between the pore solution and the silicate network was achieved. The external area, ~250-nm thick (or less, according to the caveat given in the Methods section) showed a significant increase in the 29Si/28Si, especially in the first nanometre (Supplementary Fig. 3) where the isotopic ratio goes up to 1. Moreover, the profiles of the outermost sublayer are similar at 7 and 209 days (Supplementary Fig. 3). The latter observations strongly suggest that the glass surface underwent dissolution and reprecipitation reactions, and that an equilibrium between the external surface of the glass and the bulk solution was achieved within a few days. Overall, these findings are consistent with those obtained in a far more diluted medium, favouring the hydrolysis of Si–O–M bonds (M=Si, Al and Zr), where the whole alteration layer was significantly and gradually enriched in Si supplied by the solution24. Here except at the extreme surface, almost none of the silicon atoms of the glassy network were completely hydrolysed. These observations lead to two conclusions: first, a thermodynamic equilibrium is achieved between the surface of the altered glass and the bulk solution despite the absence of isotopic equilibrium inside the porous material; second, the altered glass cannot result from the congruent dissolution of the glass. The properties of the alteration layer next needed to be determined, to better understand the behaviour of the mobile species.
The alteration layer acts as a molecular sieve
It has long been thought that Si-rich amorphous alteration layers formed during glass or mineral corrosion could be passivating25,61. The B profile was calculated with the ImageJ software.
NMR spectroscopy
Liquid-state 29Si NMR spectra were recorded on a 400-MHz Agilent DD2 spectrometer equipped with the OneNMR 5-mm probe and without sample-spinning, following the procedure described elsewhere13. 29Si cross-polarization magic-angle spinning (CP-MAS) NMR spectra were collected on a Bruker Avance 300 MHz spectrometer (magnetic field 7.05 T) using a 4-mm (rotor outer diameter) Bruker CP-MAS Probe and sample-spinning frequency of 10,000 Hz. CPMG 29Si MAS spectra collected are described in ref. 40, co-adding typically 20–40 echoes, with a recyle delay of 20 s (no changes in lineshape were observed between 2 and 200 s). CP-MAS 29Si{1H} spectra were collected with a recycle delay of 2 s and 1H-29Si magnetization transfer ranging from 0.5 to 16 ms.
Consistency of the measurements
Consistency of the glass alteration thickness measurements was verified using four independent techniques. It was found that all the measurements are consistent within a range of ±15% (Supplementary Fig. 4).
Additional information
How to cite this article: Gin, S. et al. Origin and consequences of silicate glass passivation by surface layers. Nat. Commun. 6:6360 doi: 10.1038/ncomms7360 (2015).
References
Gin, S. et al. An international initiative on long-term behavior of high-level nuclear waste glass. Mater. Today 16, 243–248 (2013).
Grambow, B. Nuclear waste glasses—how durable? Elements 2, 357–364 (2006).
Ojovan, M. I. & Lee, W. E. Glassy wasteforms for nuclear waste immobilization. Metall. Mater. Trans. A 42A, 837–851 (2011).
Walton, A. W., Schiffman, P. & Macpherson, G. L. Alteration of hyaloclastites in the HSDP 2 Phase 1 Drill Core: 2. Mass balance of the conversion of sideromelane to palagonite and chabazite. Geochem. Geophys. Geosyst. 6, Q09G19 (2005).
Sissmann, O. et al. Enhanced olivine carbonation within a basalt as compared to single-phase experiments: reevaluating the potential of CO2 mineral sequestration. Environ. Sci. Technol. 48, 5512–5519 (2014).
Vienna, J. D., Ryan, J. V., Gin, S. & Inagaki, Y. Current Understanding and remaining challenges in modeling long-term degradation of borosilicate nuclear waste glasses. Int. J. Appl. Glass Sci. 4, 283–294 (2013).
Grambow, B. & Muller, R. First-order dissolution rate law and the role of surface layers in glass performance assessment. J. Nucl. Mater. 298, 112–124 (2001).
Matzke, H. & Vernaz, E. Thermal and physicochemical properties important for the long-term behavior of nuclear waste glasses. J. Nucl. Mater. 201, 295–309 (1993).
Gin, S. et al. Nuclear glass durability: new insight into alteration layer properties. J. Phys. Chem. C 115, 18696–18706 (2011).
McGrail, B. P., Ebert, W. L., Bakel, A. J. & Peeler, D. K. Measurement of kinetic rate law parameters on a Na-Ca-Al borosilicate glass for low-activity waste. J. Nucl. Mater. 249, 175–189 (1997).
Crovisier, J. L., Advocat, T. & Dussossoy, J. L. Nature and role of natural alteration gels formed on the surface of ancient volcanic glasses (Natural analogs of waste containment glasses). J. Nucl. Mater. 321, 91–109 (2003).
Gin, S., Beaudoux, X., Angeli, F., Jegou, C. & Godon, N. Effect of composition on the short-term and long-term dissolution rates of ten borosilicate glasses of increasing complexity from 3 to 30 oxides. J. Non-Cryst. Solids 358, 2559–2570 (2012).
Gin, S. et al. The fate of silicon during glass corrosion under alkaline conditions: a mechanistic and kinetic study with the international simple glass. Geochim. Cosmochim. Acta 151, 68–85 (2015).
Beaucaire, C. et al. A thermodynamic model for the prediction of pore water composition of clayey rock at 25 and 80 degrees C—comparison with results from hydrothermal alteration experiments. Chem. Geol. 334, 62–76 (2012).
Inagaki, Y., Kikunaga, T., Idemitsu, K. & Arima, T. Initial dissolution rate of the international simple glass as a function of ph and temperature measured using microchannel flow-through test method. Int. J. Appl. Glass Sci. 4, 317–327 (2013).
Lasaga, A. C., Soler, J. M., Ganor, J., Burch, T. E. & Nagy, K. L. Chemical-weathering rate laws and global geochemical cycles. Geochim. Cosmochim. Acta 58, 2361–2386 (1994).
Grambow, B. A general rate equation for nuclear waste glass corrosion. Mater. Res. Soc. Symp. Proc. 12, 15–27 (1985).
Frugier, P. et al. SON68 nuclear glass dissolution kinetics: current state of knowledge and basis of the new GRAAL model. J. Nucl. Mater. 380, 8–21 (2008).
Daux, V., Guy, C., Advocat, T., Crovisier, J. L. & Stille, P. Kinetic aspects of basaltic glass dissolution at 90 degrees C: role of aqueous silicon and aluminium. Chem. Geol. 142, 109–126 (1997).
Gin, S., Ryan, J. V., Schreiber, D. K., Neeway, J. & Cabie, M. Contribution of atom-probe tomography to a better understanding of glass alteration mechanisms: application to a nuclear glass specimen altered 25 years in a granitic environment. Chem. Geol. 349, 99–109 (2013).
Bunker, B. C. molecular mechanisms for corrosion of silica and silicate-glasses. J. Non-Cryst. Solids 179, 300–308 (1994).
Casey, W. H. & Bunker, B. Leaching of Mineral and glass surfaces during dissolution. Rev. Mineral. Geochem. 23, 397–426 (1990).
Angeli, F. et al. Influence of calcium on sodium aluminosilicate glass leaching behaviour. Phys. Chem. Glasses 42, 279–286 (2001).
Valle, N. et al. Elemental and isotopic (29Si and 18O) tracing of glass alteration mechanisms. Geochim. Cosmochim. Acta 74, 3412–3431 (2010).
Gin, S., Ribet, I. & Couillard, M. Role and properties of the gel formed during nuclear glass alteration: importance of gel formation conditions. J. Nucl. Mater. 298, 1–10 (2001).
**ng, S. B., Buechele, A. C. & Pegg, I. L. Effect of surface-layers on the dissolution of nuclear waste glasses. Mater. Res. Soc. Symp. Proc. 333, 541–548 (1994).
Delage, F., Ghaleb, D., Dussossoy, J. L., Chevallier, O. & Vernaz, E. A mechanistic model for understanding nuclear waste glass dissolution. J. Nucl. Mater. 190, 191–197 (1992).
Daval, D. et al. Influence of amorphous silica layer formation on the dissolution rate of olivine at 90 degrees C and elevated pCO(2). Chem. Geol. 284, 193–209 (2011).
Wang, Y. F. Nanogeochemistry: nanostructures, emergent properties and their control on geochemical reactions and mass transfers. Chem. Geol. 378, 1–23 (2014).
Boksay, Z., Bouquet, G. & Dobos, S. Kinetics of formation of leached layers on glass surfaces. Phys. Chem. Glasses 9, 69–71 (1968).
Lanford, W. A. et al. Hydration of soda-lime glass. J. Non-Cryst. Solids 33, 249–266 (1979).
Doremus, R. H. in Treatise on Materials Science Vol. 17, (eds Tomozawa M., Doremus R. H. 41–69Academic Press (1979).
Ledieu, A., Devreux, F., Barboux, P., Sicard, L. & Spalla, O. Leaching of borosilicate glasses. I. Experiments. J. Non-Cryst. Solids 343, 3–12 (2004).
Angeli, F., Charpentier, T., Gin, S. & Petit, J. C. O-17 3Q-MAS NMR characterization of a sodium aluminoborosilicate glass and its alteration gel. Chem. Phys. Lett. 341, 23–28 (2001).
Cho, H. et al. Solution state structure determination of silicate oligomers by Si-29 NMR spectroscopy and molecular modeling. J. Am. Chem. Soc. 128, 2324–2335 (2006).
Iler, R. K. The Chemistry of Silica John Wiley & Sons (1979).
Engelhardt, G. & Michel, D. High-Resolution Solid-State NMR of Silicates and Zeolites John Wiley & Sons (1987).
Nanba, T., Nishimura, M. & Miura, Y. A theoretical interpretation of the chemical shift of Si-29 NMR peaks in alkali borosilicate glasses. Geochim. Cosmochim. Acta 68, 5103–5111 (2004).
Soleilhavoup, A., Delaye, J. M., Angeli, F., Caurant, D. & Charpentier, T. Contribution of first-principles calculations to multinuclear NMR analysis of borosilicate glasses. Magn. Reson. Chem. 48, S159–S170 (2010).
Angeli, F., Charpentier, T., de Ligny, D. & Cailleteauz, C. Boron speciation in soda-lime borosilicate glasses containing zirconium. J. Am. Ceram. Soc. 93, 2693–2704 (2010).
Schneider, J. et al. Q(n) distribution in stoichiometric silicate glasses: thermodynamic calculations and Si-29 high resolution NMR measurements. J. Non-Cryst. Solids 325, 164–178 (2003).
Larsen, F. H. & Farnan, I. Si-29 and O-17 (Q)CPMG-MAS solid-state NMR experiments as an optimum approach for half-integer nuclei having long T-1 relaxation times. Chem. Phys. Lett. 357, 403–408 (2002).
Dohmen, L. et al. Pattern Formation in Silicate Glass Corrosion Zones. Int. J. Appl. Glass Sci. 4, 357–370 (2013).
Geisler, T. et al. Aqueous corrosion of borosilicate glass under acidic conditions: a new corrosion mechanism. J. Non-Cryst. Solids 356, 1458–1465 (2010).
Hellmann, R. et al. Unifying natural and laboratory chemical weathering with interfacial dissolution-reprecipitation: a study based on the nanometer-scale chemistry of fluid-silicate interfaces. Chem. Geol. 294, 203–216 (2012).
Geisler, T., Schaltegger, U. & Tomaschek, F. Re-equilibration of zircon in aqueous fluids and melts. Elements 3, 43–50 (2007).
McGrail, B. P. et al. The structure of Na2O-Al2O3-SiO2 glass: impact on sodium ion exchange in H2O and D2O. J. Non-Cryst. Solids 296, 10–26 (2001).
Cailleteau, C. et al. Insight into silicate-glass corrosion mechanisms. Nat. Mater. 7, 978–983 (2008).
Jollivet, P. et al. Investigation of gel porosity clogging during glass leaching. J. Non-Cryst. Solids 354, 4952–4958 (2008).
Bourg, I. C. & Steefel, C. I. Molecular dynamics simulations of water structure and diffusion in silica nanopores. J. Phys. Chem. C 116, 11556–11564 (2012).
Briman, I. M. et al. Impact of pore size and pore surface composition on the dynamics of confined water in highly ordered porous silica. J. Phys. Chem. C 116, 7021–7028 (2012).
Chamssedine, F., Sauvage, T., Peuget, S., Fares, T. & Martin, G. Helium diffusion coefficient measurements in R7T7 nuclear glass by He-3(d,alpha)H-1 nuclear reaction analysis. J. Nucl. Mater. 400, 175–181 (2010).
Libourel, G. et al. The use of natural and archeological analogues for understanding the long-term behavior of nuclear glasses. C. R. Geosci. 343, 237–245 (2011).
Michelin, A. et al. Silicate glass alteration enhanced by iron: origin and long-term Implications. Environ. Sci. Technol. 47, 750–756 (2013).
Curti, E., Crovisier, J. L., Morvan, G. & Karpoff, A. M. Long-term corrosion of two nuclear waste reference glasses (MW and SON68): a kinetic and mineral alteration study. Appl. Geochem. 21, 1152–1168 (2006).
Cunnane, J. C. et al. High-level nuclear-waste borosilicate glass—a compendium of characteristics. Mater. Res. Soc. Symp. Proc. 294, 225–232 (1993).
Bourcier, W. L., Peiffer, D. W., Knauss, K. G., Mckeegan, K. D. & Smith, D. K. A kinetic-model for borosilicate glass dissolution based on the dissolution affinity of a surface alteration layer. Mater. Res. Soc. Symp. Proc. 176, 209–216 (1990).
Munier, I., Crovisier, J. L., Grambow, B., Fritz, B. & Clement, A. Modelling the alteration gel composition of simplified borosilicate glasses by precipitation of an ideal solid solution in equilibrium with the leachant. J. Nucl. Mater. 324, 97–115 (2004).
Crank, J. The Mathematics of Diffusion Oxford Science Publication (1980).
Giannuzzi, L. A. & Stevie, F. A. A review of focused ion beam milling techniques for TEM specimen preparation. Micron 30, 197–204 (1999).
Bonnet, N., Colliex, C., Mory, C. & Tence, M. Developments in processing image sequences for elemental map**. Scanning Microsc. 2, 351–364 (1988).
Acknowledgements
This work was supported by CEA and Areva. We are grateful to Yves Minet, Alex Mitroshkov, Laurent Dupuy, Claude Berthon, Martiane Cabié, Jean-Louis Chouchan, Céline Marcou, Jessica Vincent and Jean-Pierre Mestre for their technical support. We also thank Baptiste Gault, Lindsey Neill, Benjamin Parruzot and François Sudreau for their helpful comments.
Author information
Authors and Affiliations
Contributions
S.G. supervised the study and wrote the paper. P.J. and M.F. were deeply involved in the interpretation. P.F. modeled the B profiles. F.A. and T.C. performed the solid-state NMR study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-4 and Supplementary Table 1 (PDF 321 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gin, S., Jollivet, P., Fournier, M. et al. Origin and consequences of silicate glass passivation by surface layers. Nat Commun 6, 6360 (2015). https://doi.org/10.1038/ncomms7360
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms7360
- Springer Nature Limited
This article is cited by
-
Effect of structural disorder induced by external irradiation with heavy ions on the alteration of a four oxide borosilicate glass
npj Materials Degradation (2024)
-
The corrosion mechanism of lead-glazed pottery in Han dynasty
npj Materials Degradation (2024)
-
Corrosion of glaze in the marine environment: study on the green-glazed pottery from the Southern Song “Nanhai I” shipwreck (1127–1279 A.D.)
Heritage Science (2023)
-
Alteration of medieval stained glass windows in atmospheric medium: review and simplified alteration model
npj Materials Degradation (2023)
-
Borosilicate glass alteration in vapor phase and aqueous medium
npj Materials Degradation (2022)