
Abstract
Attention-deficit/hyperactivity disorder (ADHD) is a common, highly heritable neurodevelopmental disorder. Genetic loci have not yet been identified by genome-wide association studies. Rare copy number variations (CNVs), such as chromosomal deletions or duplications, have been implicated in ADHD and other neurodevelopmental disorders. To identify rare (frequency ⩽1%) CNVs that increase the risk of ADHD, we performed a whole-genome CNV analysis based on 489 young ADHD patients and 1285 adult population-based controls and identified one significantly associated CNV region. In tests for a global burden of large (>500 kb) rare CNVs, we observed a nonsignificant (P=0.271) 1.126-fold enriched rate of subjects carrying at least one such CNV in the group of ADHD cases. Locus-specific tests of association were used to assess if there were more rare CNVs in cases compared with controls. Detected CNVs, which were significantly enriched in the ADHD group, were validated by quantitative (q)PCR. Findings were replicated in an independent sample of 386 young patients with ADHD and 781 young population-based healthy controls. We identified rare CNVs within the parkinson protein 2 gene (PARK2) with a significantly higher prevalence in ADHD patients than in controls (P=2.8 × 10−4 after empirical correction for genome-wide testing). In total, the PARK2 locus (chr 6: 162 659 756–162 767 019) harboured three deletions and nine duplications in the ADHD patients and two deletions and two duplications in the controls. By qPCR analysis, we validated 11 of the 12 CNVs in ADHD patients (P=1.2 × 10−3 after empirical correction for genome-wide testing). In the replication sample, CNVs at the PARK2 locus were found in four additional ADHD patients and one additional control (P=4.3 × 10−2). Our results suggest that copy number variants at the PARK2 locus contribute to the genetic susceptibility of ADHD. Mutations and CNVs in PARK2 are known to be associated with Parkinson disease.
Similar content being viewed by others

Introduction
Attention-deficit/hyperactivity disorder (ADHD) represents one of the most common psychiatric disorders in children and adolescents with a worldwide prevalence rate of 5.2%.1 According to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV),2 ADHD is characterized by pervasive and impairing symptoms of inattention, hyperactivity and increased impulsivity. Family, twin and adoption studies indicate that ADHD is a highly heritable disorder; heritability estimates are consistently around 0.8.3, 4, 5, 6, 7 However, neither genome-wide association studies (GWAS) nor large scale meta-analyses of GWAS has so far unequivocally identified specific genes conferring major risk (see Hinney et al.8).
Copy number variations (CNVs) are, by definition, chromosomal deletions or duplications of at least 1 kb up to several Mb that are variable in size among carriers. At a genome-wide level, thousands of CNVs have already been identified. Several CNVs were shown to contribute to neurodevelopmental disorders such as schizophrenia,9 Parkinson disease (PD)10 or autism.11 Five genome-wide analyses of CNVs have been published in ADHD.Supplementary Text. All association analyses were performed on the QC filtered, rare CNV calls were investigated using the PLINK software (version 1.07).30 As primary analyses, we tested the hypothesis that particular rare CNVs might be found at an increased frequency in ADHD cases compared with controls. Locus-specific tests of association were performed (one-sided χ2 tests) and significance was assessed via permutation (empirical P-values based on 100 000 permutations) at a pointwise as well as at a genome-wide level. In more detail, we calculated the frequency of rare CNVs in ADHD patients, and we compared it to the frequency in the controls. The frequency was calculated at each unique start and stop site for rare CNVs that met all of the defined QC measures (defined in the Supplementary Text). Each site (5047 sites in total, located in 1083 non-overlap** genomic CNV containing regions) was assessed for a difference in CNV frequency between groups with the use of a permutation-based Fisher’s exact test in PLINK. We refer to a locus in the sense of a susceptibility locus as a genomic region that is exclusively made up of adjacently tested sites for which significantly more rare CNVs were observed in ADHD cases than in controls. These analyses were undertaken for all rare CNVs as well as stratified according to CNV type, that is, deletion or duplication. In the GWAS sample, empirical, genome-wide corrected P-values were generated by permuting affection status and simultaneously preserving the correlation structure of CNVs (100 000 permutations) to simulate the null hypothesis of no association. In other words, we used the permutation resampling method to correct for the multiple testing problem, which occurs when testing any identified locus of rare CNVs.
By application of PLINK’s case–control ‘cnv-enrichment-test’ function, we additionally tested whether CNVs in the PARK2 gene are enriched in ADHD GWAS cases compared with GWAS controls. In contrast to Fisher’s exact test, the ‘cnv-enrichment-test’ is robust to case–control differences in CNV size or CNV rate.31 Enrichment in cases is reported as one-sided empirical P-value using 100 000 permutations.
Analysis of the replication sample was performed to confirm the finding of the GWAS sample at the PARK2 locus (Results section) and we focussed the testing on this single rare CNV locus for an association to ADHD. Consequently, pointwise empirical P-values (100 000 permutations) for the replication sample were not corrected for multiple testing. The statistical analyses in the GWAS sample were repeated for quantitative (q)PCR-validated CNVs at the PARK2 locus as part of the sensitivity analysis. We applied a significance level α of 0.05 (globally for the genome-wide testing and locally for the replication sample).
As secondary sensitivity analyses (see Supplementary Text), we also assessed the genome-wide frequency of CNVs in ADHD cases compared with controls according to the average number of CNVs per sample. We expected more CNVs in the ADHD cases based on the literature.32 Thus, one-sided tests were applied to all rare CNVs, as well as to rare deletions and duplications only; and genome-wide multiple testing was dealt with using 100 000 permutations. Finally, we likewise tested whether CNVs in the ADHD cases were larger in size than those in the control group based on the average size of CNVs per individual.
CNV validation and replication analysis at the PARK2 locus
We performed real-time qPCR experiments to validate the CNVs by a Duplex TaqMan CNV assay (Applied Biosystems, Darmstadt, Germany, assay Hs03615859_cn at chr6: 162 696 987±50 bp, NCBI36/hg18) as described previously.33 Individual copy number status was determined for each ADHD patient of the GWAS sample. Briefly, every PCR was performed as a triplicate for each individual of the GWAS ADHD cases and the results from the qPCR were analysed using the software CopyCaller 1.0 (Applied Biosystems). In cases, 11 of the 12 CNVs identified with PennCNV covering the PARK2 locus (Results section) were technically validated by qPCR. qPCR experiments did not reveal any further CNV carrier, which was undetected in previous SNP array-based CNV detection analyses. Thus, CNVs at the PARK2 locus of GWAS ADHD patients could be validated with both, a low false-positive and a low false-negative rate. For a subset of controls (HNR controls), CNV calls, which were estimated to cover the PARK2 gene, were validated by qPCR. Apart from one potential CNV carrier, who was incorporated into statistical analysis at the PARK2 locus, we additionally considered the five HNR controls for which CNVs were estimated to flank the PARK2 locus (n=3) or for which CNVs were called but excluded due to their small size (spanned <15 probes) in the course of our CNV QC procedure (n=2). Moreover, we additionally included six randomly chosen control subjects of the HNR control sample. CNV analyses were performed blinded to the likely CNV status of the controls. For the PARK2 locus, all analysed CNV states could be validated. For the KORA and PopGen controls, no DNA was available for qPCR validation. However, given the high technical validation rate in the available DNA samples, validity of CNV calls was presumed to be comparably high for KORA and PopGen controls. Although false-negative and false-positive rates are unknown for the GWAS controls group, there is no obvious reason to expect that these rates would significantly differ between cases and controls. Notably, the low frequency of PARK2 CNVs in control subjects was consistently reported in the ‘Database of Genomic Variants’ (http://projects.tcag.ca/variation) and in two previous publications, where CNVs at the PARK2 gene were absent in 2026 healthy, population-based controlsSupplementary Text.
Results
The GWAS sample included 489 ADHD cases and 1285 controls (Table 1) with high-quality SNP array data for full CNV analysis. Comparison of the CNV sets identified in the ADHD patients and in the controls showed no increased overall frequency of CNVs in ADHD cases (Supplementary Text). After exclusion of common (frequency >1%) CNVs, 2432 rare CNVs (592 in ADHD cases; 1840 in controls) with an increased length in ADHD cases (average CNV size: 226.3 kb (range: 9.3–2830.8 kb) in ADHD cases; 186.4 kb (range: 5.6–4479.6 kb) in controls) were included in the association analysis. Although there was a difference in the sex distribution between ADHD patients (81.0% males) and control subjects (50.7% males), there was no evidence for significant difference in the rate at which rare CNVs were called in males compared with females in either cases or comparison subjects (data not presented). All rare CNVs >500 kb are listed in Supplementary Table S1.
With regard to previous observations,13, 16, 17 we first looked at our data with respect to a potential overall enrichment of rare CNVs in ADHD cases compared with controls (Supplementary Table S7) and in terms of an enrichment for loci implicated in neurodevelopmental disorders, such as autism or schizophrenia (data not shown). There was no evidence for an increased burden of rare CNVs in ADHD patients (P=0.997). We additionally performed comparative analyses on rare CNVs stratified by their size. Interestingly, with increasing size thresholds, we observed a stronger trend of association between large, rare CNVs and ADHD, which is in accordance with the reports of previous studies.13, 16 Despite the fact that none of the comparisons resulted in a nominally significance (that is, P<0.05): there was a 1.126-fold enriched rate (P=0.271) and a 1.133-fold higher proportion (P=0.253) of subjects carrying at least one rare CNV >500 kb in the ADHD cohort. The rate of rare CNVs >500 kb observed in ADHD cases was 10.4%, which is similar to the rates of 12.2 and 12.5% reported in previous studies.13, 16 Limiting our analysis to rare CNVs >2 Mb, we observed a 3.065-fold enrichment (P=0.074) in ADHD cases relative to control subjects. However, due to the potential bias in individual CNV rate and average size, which differentiates cases and controls in our GWAS sample (see Supplementary Text), we did not follow-up these data. Differences between distributions in cases and controls may rather result from different technical genoty** procedures, than indicating association effects.
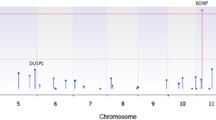
Locus-specific association tests for an overrepresentation of CNVs, including both deletions and duplications, in ADHD cases in comparison with controls revealed only one genome-wide significant genomic region within the PARK2 gene with a P-value of 2.8 × 10−4 empirically corrected for genome-wide testing (Figure 1, Supplementary Table S8). This locus for which we observed more rare CNVs in ADHD cases than in controls is located at chr6: 162 659 756—162 767 019 (NCBI36/hg18). We refer to this region as the PARK2 locus. In total, this locus is covered by 12 CNVs among the ADHD patients (2.45%, three deletions (0.61%) and nine duplications (1.84%)) and four CNVs among the controls (0.31%, two deletions (0.16%) and two duplications (0.16%)), all these CNVs extend into the coding region (either exon 2 or exon 3) of PARK2. Locus-specific association tests, stratified according to CNV type (deletion or duplication), did not reveal further genomic regions with genome-wide significant results. The PARK2 locus alone showed a genome-wide significant enrichment of duplications in ADHD cases compared with controls (P=1.9 × 10−3 after empirical correction for genome-wide testing, Figure 1). In contrast, we did not observe a genome-wide significant excess of deletions for any of the tested regions harbouring rare CNVs.

Results for the PARK2 locus in the GWAS and in the replication sample. Each panel consists of four parts (called CNVs, PARK2 gene, probes analysed and association tests): CNVs: red (pink) bars represent duplications in an ADHD case (control), blue (lightblue) bars indicate a deletion of an ADHD case (control). PARK2 gene: the marks indicate the coding regions (NCBI36/hg18). Association tests: permutation-based one-sided −log10-transformed P-values for association tests; the black (pink; lightblue) line represents association tests for an increased frequency of segmental CNV data independent of type (deletions; duplications) in cases compared with controls. The significance level P=0.05 is highlighted as a dashed red line. The chromosomal region offering genome-wide significantly more CNVs in ADHD patients than in controls is highlighted by grey vertical shading. (a) Results for the GWAS sample. The presented P-values are genome-wide empirically corrected. The chromosomal region covered by the qPCR assay used for validation of PennCNV’s CNV calls is shown as a darkgrey vertical dashed line within this region of genome-wide significance. The duplication that could not be validated by qPCR analysis is marked by ‘x’. Results of association tests after exclusion of the non-validated case duplication are shown in Supplementary Figure S7. (b) Results for the replication sample. The presented P-values are pointwise corrected.
Enrichment of CNVs at the PARK2 gene for GWAS ADHD cases compared with GWAS controls was also supported by the robust ‘cnv-enrichment-test’31 (one-sided empirical P=8.8 × 10−4).
Copy number status at the PARK2 locus of all ADHD patients in the GWAS sample was reevaluated by qPCR analyses (Figure 1). Apart from one duplication, each CNV status was technically validated. Even after reanalysis of all rare CNVs with exclusion of the non-validated duplication, the finding for the PARK2 locus remained genome-wide significant (empirically corrected P=1.2 × 10−3, Supplementary Figure S7). We observed no differences in ADHD subtypes or basic characteristics like age, sex and intelligence quotient (IQ) values between carriers of CNVs at the PARK2 locus and the 489 ADHD patients of the GWAS sample (Supplementary Table S2).
Next, we assessed an independent sample of 386 ADHD patients and 781 healthy controls to replicate our finding of an excess of CNVs in ADHD patients at the PARK2 locus. We replicated the excess (P=4.3 × 10−2, Figure 1) of CNVs in the ADHD replication sample (n=4 (1.04%), two duplications (0.52%) and two deletions (0.52%)) compared with replication controls (one duplication (0.13%)). Similar to the initial finding, four of the five CNVs in the PARK2 locus extend into the coding exon 2 of the gene. Owing to the small number of CNVs we did not stratify by CNV status.
Finally, in addition to our GWAS discovery analysis, we assessed ADHD CNV candidate loci from previous reportsSupplementary Text) may in part be explained by the inability of a precise determination of CNV breakpoints on the basis of SNP chip data. To support our main finding, however, the association of PARK2 CNVs with ADHD was validated by qPCR and replicated in an independent sample.
In summary, our results support the role of structural variants at the PARK2 locus for ADHD genetics. Moreover, our data support the further investigation of CNVs involving neurodevelopmental genes, such as CHL1, PTPRD, GCNT2, PTPRN2 and NDE1, as well as deletions and duplications at the 15q13 and 16p11.2 regions for ADHD genetics.
References
Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA . The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 2007; 164: 942–948.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Diseases (DSM-IV) 4th ed. American Psychiatric Publishing: Washington, DC, 1994.
Heiser P, Friedel S, Dempfle A, Konrad K, Smidt J, Grabarkiewicz J et al. Molecular genetic aspects of attention-deficit/hyperactivity disorder. Neurosci Biobehav Rev 2004; 28: 625–641.
Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA et al. Molecular genetics of attention deficit/hyperactivity disorder. Biol Psychiatry 2005; 57: 1313–1323.
Faraone SV, Mick E . Molecular genetics of attention deficit hyperactivity disorder. Psychiatr Clin North Am 2010; 33: 159–180.
Franke B, Neale BM, Faraone SV . Genome-wide association studies in ADHD. Hum Genet 2009; 126: 13–50.
Freitag CM, Rhode LA, Lempp T, Romanos M . Phenotypic and measurement influences on heritability estimates in childhood ADHD. Eur Child Adolesc Psychiatry 2010; 19: 311–323.
Hinney A, Scherag A, Jarick I, Albayrak Ö, Pütter C, Pechlivanis S et al. Genome-wide association study in German patients with attention deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2011; 156: 888–897.
Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008; 455: 232–236.
Pankratz N, Dumitriu A, Hetrick KN, Sun M, Latourelle JC, Wilk JB et al. Copy number variation in familial parkinson disease. PLoS One 2011; 6: e20988.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009; 459: 569–573.
Elia J, Gai X, **e HM, Perin JC, Geiger E, Glessner JT et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry 2010; 15: 637–646.
Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376: 1401–1408.
Lesch KP, Selch S, Renner TJ, Jacob C, Nguyen TT, Hahn T et al. Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol Psychiatry 2011; 16: 491–503.
Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med 2011; 3: 95ra75.
Williams NM, Franke B, Mick E, Richard JL, Freitag CM, Gill M et al. Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13.3. Am J Psychiatry 2012; 169: 195–204.
Stergiakouli E, Hamshere M, Holmans P, Langley K, Zaharieva I et aldeCODE Genetics. Investigating the contribution of common genetic variants to the risk and pathogenesis of ADHD. Am J Psychiatry 2012; 169: 186–194.
Elia J, Glessner JT, Wang K, Takahashi N, Shtir CJ, Hadley D et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet 2011; 44: 78–84.
Mayo O . The rise and fall of the common disease-common variant (CD-CV) hypothesis: how the sickle cell disease paradigm led us all astray (or did it?). Twin Res Hum Genet 2007; 10: 793–804.
Hebebrand J, Dempfle A, Saar K, Thiele H, Herpertz-Dahlmann B, Linder M et al. A genome-wide scan for attention-deficit/hyperactivity disorder in 155 German sib-pairs. Mol Psychiatry 2006; 11: 196–205.
Schimmelmann BG, Friedel S, Dempfle A, Warnke A, Lesch KP, Walitza S et al. No evidence for preferential transmission of common valine allele of the Val66Met polymorphism of the brain-derived neurotrophic factor gene (BDNF) in ADHD. J Neural Transm 2007; 114: 523–526.
Romanos M, Freitag C, Jacob C, Craig DW, Dempfle A, Nguyen TT et al. Genome-wide linkage analysis of ADHD using high-density SNP arrays: novel loci at 5q13.1 and 14q12. Mol Psychiatry 2008; 13: 522–530.
Cichon S, Mühleisen TW, Degenhardt FA, Mattheisen M, Miró X, Strohmaier J et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet 2011; 88: 372–381.
Schmermund A, Möhlenkamp S, Stang A, Grönemeyer D, Seibel R, Hirche H et al. Assessment of clinically silent atherosclerotic disease and established and novel risk factors for predicting myocardial infarction and cardiac death in healthy middle-aged subjects: rationale and design of the Heinz Nixdorf RECALL Study. Risk factors, evaluation of coronary calcium and lifestyle. Am Heart J 2002; 144: 212–218.
Krawczak M, Nikolaus S, von Eberstein H, Croucher PJ, El Mokhtari NE, Schreiber S . PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet 2006; 9: 55–61.
Wichmann HE, Gieger C, Illig T, Group, MONICA/KORA Study. KORA-gen—resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen 2005; 67: S26–S30.
Renner TJ, Walitza S, Dempfle A, Eckert L, Romanos M, Gerlach M et al. Allelic variants of SNAP25 in a family-based sample of ADHD. J Neural Transm 2008; 115: 317–321.
Zutavern A, Brockow I, Schaaf B, Bolte G, von Berg A, Diez U et al. Timing of solid food introduction in relation to atopic dermatitis and atopic sensitization: results from a prospective birth cohort study. Pediatrics 2006; 117: 401–411.
Berg A, Kramer U, Link E, Bollrath C, Heinrich J, Brockow I et al. Impact of early feeding on childhood eczema: development after nutritional intervention compared with the natural course—the GINIplus study up to the age of 6 years. Clin Exp Allergy 2010; 40: 627–636.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Raychaudhuri S, Korn JM, McCarroll SA, International Schizophrenia Consortium, Altshuler D, Sklar P et al. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet 2010; 6, pii: e1001097.
Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, Vu TH et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet 2011; 7: e1002334.
deKovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2010; 133: 23–32.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008; 358: 667–675.
Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet 2008; 40: 1466–1471.
Xu B, Roos JL, Levy S, van Rensburg EJ, Gogos JA, Karayiorgou M . Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet 2008; 40: 880–885.
Jiang H, Jiang Q, Feng J . Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J Biol Chem 2004; 279: 54380–54386.
Crosiers D, Theuns J, Cras P, Van Broeckhoven C . Parkinson disease: insights in clinical, genetic and pathological features of monogenic disease subtypes. J Chem Neuroanat 2011; 42: 131–141.
Chien HF, Rohé CF, Costa MD, Breedveld GJ, Oostra BA, Barbosa ER et al. Early-onset Parkinson’s disease caused by a novel parkin mutation in a genetic isolate from north-eastern Brazil. Neurogenetics 2006; 7: 13–19.
Walitza S, Melfsen S, Herhaus G, Scheuerpflug P, Warnke A, Müller T et al. Association of Parkinson’s disease with symptoms of attention deficit hyperactivity disorder in childhood. J Neural Transm 2007; 72 (Suppl): 311–315.
Langley K, Martin J, Agha SS, Davies C, Stergiakouli E, Holmans P et al. Clinical and cognitive characteristics of children with attention-deficit hyperactivity disorder, with and without copy number variants. Br J Psychiatry 2011; 199: 398–403.
Bradshaw NJ, Christie S, Soares DC, Carlyle BC, Porteous DJ, Millar JK . NDE1 and NDEL1: multimerisation, alternate splicing and DISC1 interaction. Neurosci Lett 2009; 449: 228–233.
Ullmann R, Turner G, Kirchhoff M, Chen W, Tonge B, Rosenberg C et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat 2007; 28: 674–682.
Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P et al. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet 2009; 18: 1497–1503.
Sakurai K, Migita O, Toru M, Arinami T . An association between a missense polymorphism in the close homologue of L1 (CHL1, CALL) gene and schizophrenia. Mol Psychiatry 2002; 7: 412–415.
Yang Q, Li L, Yang R, Shen GQ, Chen Q, Foldvary-Schaefer N et al. Family-based and population-based association studies validate PTPRD as a risk factor for restless legs syndrome. Mov Disord 2011; 26: 516–519.
van der Zwaag B, Franke L, Poot M, Hochstenbach R, Spierenburg HA, Vorstman JA et al. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS One 2009; 4: e5324.
Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 2010; 47: 332–341.
Miller DT, Shen Y, Weiss LA, Korn J, Anselm I, Bridgemohan C et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet 2009; 46: 242–248.
Acknowledgements
We thank the children and their families for their participation and support to this study. We are also grateful to all probands from the community-based cohorts of PopGen, KORA, those from the Heinz Nixdorf RECALL (HNR) study, and the GINIplus and LISAplus cohorts. We thank the Heinz Nixdorf Foundation, Germany, for the generous support of the HNR study. We thank the German Research Association (DFG) who funded the GWAS analyses and confirmatory studies (He1446/9-1 to J Hebebrand, KP Lesch, A Hinney and T Renner, KFO 125, SFB 581, SFB TRR 58/A5, GRK 1253 to KP Lesch; ME 1923/5-1, ME 1923/5-3 to J Meyer and CM Freitag, GRK 1389 to J Meyer, SCHA 542/10-3 to H Schäfer) and the Bundesministerium für Bildung und Forschung (BMBF 01GV0605 to KP Lesch). We thank the START-Program EK 119/05 of the Medical Faculty, RWTH Aachen, Germany. The European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 245009 supported this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
KH Jöckel and A Scherag are responsible statisticians in several clinical trials and in this position they receive support money from various pharmaceutical companies, among them MEDICE Arzneimittel Pütter GmbH and KG. In the past year, Dr Faraone received consulting income and research support from Shire and Alcobra and research support from the National Institutes of Health. In previous years, he received consulting fees or was on Advisory Boards or participated in continuing medical education programs sponsored by: Shire, McNeil, Janssen, Novartis, Pfizer and Eli Lilly. Dr Faraone receives royalties from books published by Guilford Press: Straight Talk about Your Child's Mental Health and Oxford University Press: Schizophrenia: The Facts. S Walitza recieves industry funding research from Vifor Pharma, Switzerland and was on the speakers' bureau of Eli Lilly, Jannssen-Cilag and Astra Zeneca. SW receives research funding from DFG, SNF, BMFFSJ and FP7. CM Freitag was on the speakers' bureau of Eli Lilly and Novartis, and was an expert consultant for Desitin. Dr Herpertz-Dahlmann receives industry research funding from Vifor Pharma, Schweiz. In previous years, she was on the Advisory Board of Eli Lilly and received sponsoring by AstraZeneca, Eli Lilly, Novartis and Janssen Cilag. She received research support from the Federal Ministry of Education and Research and the German Research Society (Deutsche Forschungsgemeinschaft). G Lehmkuhl receives industry research funding from Lilly Deutschland GmbH, Germany. A Warnke recieves industry research funding from Shire and in previous years from Eli Lilly. B Schimmelmann is a member of the speakers' bureau of Eli Lilly, BMS, Janssen and Novartis. The other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Jarick, I., Volckmar, AL., Pütter, C. et al. Genome-wide analysis of rare copy number variations reveals PARK2 as a candidate gene for attention-deficit/hyperactivity disorder. Mol Psychiatry 19, 115–121 (2014). https://doi.org/10.1038/mp.2012.161
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2012.161
- Springer Nature Limited
Keywords
This article is cited by
-
Rare copy number variants in males and females with childhood attention-deficit/hyperactivity disorder
Molecular Psychiatry (2023)
-
A comprehensive analysis of copy number variation in a Turkish dementia cohort
Human Genomics (2021)
-
Genetic variations influence brain changes in patients with attention-deficit hyperactivity disorder
Translational Psychiatry (2021)
-
Copy Number Variants and Polygenic Risk Scores Predict Need of Care in Autism and/or ADHD Families
Journal of Autism and Developmental Disorders (2021)
-
Mitochondrial DNA haplogroups and risk of attention deficit and hyperactivity disorder in European Americans
Translational Psychiatry (2020)