
Abstract
Pyruvate dehydrogenase E1-alpha deficiency (PDHAD) results in lactic acidosis and hyperpyruvatemia. Two patients with PDHAD, a man with a p.R263Q mutation, and a girl with a p.C145del mutation in PDHE1α, presented with lactic acidosis with neurological disorder. These patients were able to survive for a long period under careful nursing care. Herein, we discuss the factors contributing to their relatively stable clinical course, albeit with intellectual disability.
Similar content being viewed by others


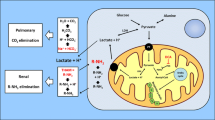
Pyruvate dehydrogenase (PDH) catalyzes the irreversible decarboxylation of pyruvate into acetyl-CoA, linking glycolysis to the tricarboxylic acid cycle. PDH is composed of multiple copies of three subunits, that is, PDH (E1, EC 1.2.4.1), dihydrolipoamide transacetylase (E2, EC 2.3.1.12) and dihydrolipoamide dehydrogenase (E3, EC 1.8.1.4), along with an E3-binding protein. The E1 component is a heterotetramer of two α-subunits and two β-subunits. The gene encoding the E1α subunit (PDHA1, MIM #300502) is located on the X chromosome (Xp22.1–22.2), and all subunits are nuclear-encoded. PDH E1α deficiency (PDHAD, MIM #312170) results in lactic acidosis and hyperpyruvatemia. The spectrum of the clinical manifestations of PDHAD ranges from severe neonatal lactic acidosis with early death to intermittent ataxia with a late-onset progressive neurodegenerative course.1,2
We followed two PDHAD patients with intellectual disability and a relatively stable clinical course under careful nursing care. We previously reported the onset and diagnosis of patient 1,3,4 whereas patient 2 presents a novel mutation (c.433_435delTGT, p.C145del mutation; http://www.hgmd.cf.ac.uk). We report the relationship between their genetic backgrounds and long-term outcomes.
Patient 1 is a 38-year-old man, who had been born at full term with a birth weight of 2,670 g. He could support his head at 6 months and could stand with support at 14 months. He first presented with apnea at 19 months and was referred to our institution. He presented with severe metabolic acidosis (blood pH, 7.30; HCO3−, 7.7 mEq/l; base excess, −15.3 mEq/l), hyperlactacidemia (50.8 mg/dl) and hyperpyruvic acidemia (3.4 mg/dl). Lactate (31 mg/dl) and pyruvate (2.1 mg/dl) levels were elevated in his cerebrospinal fluid (CSF). Brain CT revealed microcephaly and a low-density area on both sides of the basal ganglia. At 21 months, he was diagnosed with a c.G788A de novo mutation in PDHA1 exon 8 (NM_000284.3), resulting in an amino acid substitution (p.R263Q). He was treated with vitamins B1 and B6. He was repeatedly hospitalized (two to four times per year) during his childhood because of convulsions and infections. His body condition gradually deteriorated at every hospitalization; he could not walk and presented with repeated aspiration pneumonia. He underwent gastrostomy at 28 years of age and laryngo-tracheal resection at 29 years of age. Thereafter, he had fewer convulsions and was never hospitalized again because further pneumonia was prevented. He lives at home with his guardian without respiratory support.
Patient 2 is a 10-year-old girl, the first child of nonconsanguineous parents. She presented with intrauterine growth restriction at 9 months of gestation. She was delivered by urgent caesarean section at 35 weeks of gestation because of preeclampsia. Her birth weight was 1,716 g. She presented mild metabolic acidosis (blood pH, 7.36; HCO3−, 18.0 mEq/l; base excess, −6.2 mEq/l). Head ultrasonography indicated ventricular enlargement on both sides and periventricular leukomalacia (Figure 1a,b). Her brain MRI at 31 days of age showed symmetrical low-intensity signals of the basal ganglia and corpus callosum hypoplasia (Figure 1c). PDHAD was suspected because of elevated blood lactate (64.7 mg/dl) and pyruvate (5.0 mg/dl) and lactate in the CSF (58.4 mg/dl) (Supplementary Table S1). She was diagnosed with a c.433_435delTGT heterozygous mutation in PDHA1 exon 5 (NM_000284.3) (Figure 1d–f), confirmed to be de novo. She was treated with vitamin B1, biotin, lipoate and dichloroacetic acid (DCA). Although hyperlactacidemia and hyperpyruvic acidemia improved to 18.4 mg/dl and 1.78 mg/dl, respectively, lactate levels in the CSF (44.1 mg/dl) had not decreased 12 days after administration. As the ventricular enlargement and brain atrophy progressed with time, DCA was discontinued at 3 years of age. She was hospitalized three times because of decreased appetite, convulsions and aspiration pneumonia. She underwent cardioplasty at 4 years of age because her gastroesophageal reflux became severe. She then demonstrated good nutritional status and did not develop aspiration pneumonia again. She is now 10 years old and has a developmental quotient of <20 on the Enjoji scale of infant analytical development and an unmeasurable intelligence quotient. She is in a stable physical state and goes to a special school.

Brain imaging and PDHA1 DNA sequence in patient 2. Brain ultrasonography ((a) coronal section and (b) sagittal section images) demonstrated enlarged lateral ventricles on both sides and cystic lesions. (c) Brain MRI showed enlarged lateral ventricles on both sides and cystic changes around the lateral ventricles. (d) Direct PDHA1 DNA sequencing of white blood cell DNA in patient 2. Subcloning of PCR products generated from white blood cell DNA from patient 2 demonstrated a mixture of the normal sequence (e) and the c.433_435delTGT (p.C145del) mutation (f).
This case report was approved by the Ethics Committee of the Faculty of Life Science, Kumamoto University. Written informed consent was obtained from the families of both patients reported herein.
No report has been published regarding patients with PDHAD who have survived as long as patient 1. Although patient 2 has fetal-onset type PDHAD, she presents with a relatively stable health condition. Patel et al.5 reported that 243 of 371 patients with PDHAD (65.5%) were alive at 6 months of age, but only 10 patients (2.7%) were still alive at 20 years of age. Death was associated with high blood lactate levels and low PDH activity. The common PDHA1 mutations are at amino acid position 263, 302 or 378. Some patients with c.R263G survive for 7–18 years.6,7, 8 Patient 1 presented with p.R263Q and has survived for 38 years to date. The enzyme activity is variable between male patients with p.R263G and p.R263Q (16–77%; Table 1). According to Barnerias et al.9 and DeBrosse et al.,10 within specimens from the same individual with PDHA1 mutations and even within the same specimen, there are large variations in PDH activity. This was evident when PDH activity was compared between cultured skin fibroblasts and fresh blood lymphocytes from the same individual. DeBrosse et al.10 reported a significant difference in the survival of males with PDHA1 mutations whose fibroblast PDH activity was ⩾35% of the reference mean compared with those whose PDH activity was <35% of the mean. However, there was no significant correlation between PDH activity in fibroblasts from male or female patients with PDHA1 mutations and their intellectual outcome. Therefore, male subjects with PDHA1 mutations whose fibroblast PDH activity is ⩾35% are more likely to have long-term survival, similar to patient 1 reported here.
The c.433_435delTGT (p.C145del) identified in patient 2 has never been reported. Gene mutations around amino acid 145 in exon 5 are summarized in Table 1. Patients with p.R141Q or p.G144del present residual activity, and the activity of p.C145del might be relatively high, although this information is not available. However, investigating PDH activity remains difficult, particularly in female patients with PDHA1 mutations, because of tissue-dependent X-inactivation. The defect might need to be sought and confirmed in several tissues by techniques such as enzymatic activity assay and immnohistochemistry.11,12, 13, Tissue-specific skewed X-chromosome inactivation gives rise to the variability of female PDHAD.
The effective treatment for PDHAD is a ketogenic diet with carbohydrate restriction. Patients 1 and 2 consumed a ketogenic diet. Biotin was administered to both patients to activate their pyruvate carboxylase, which catalyzes pyruvate carboxylation to form oxaloacetate. Moreover, vitamin B1 and DCA might be useful. DCA was only administered to patient 2. DCA decreases lactate and pyruvate levels in the blood and CSF by inhibiting PDH kinase and stabilizing the PDH complex. After DCA administration in patient 2, lactate levels decreased in the blood, but not in the CSF at 12 days after administration. Neurological symptoms gradually improve from 1 to 24 months after administration of DCA.14 Thus, improvement in CSF and neurological symptoms might be observable after at least a month of DCA administration.
Although patient 1 achieved head control late, he underwent a detailed examination for PDHAD as late as at 19 months. Intervention at an earlier stage might have improved the outcome. He was repeatedly hospitalized during his childhood because of convulsions and infections, and his general physical condition deteriorated every time he contracted an infection. Moreover, his general medical condition deteriorated further after receiving dextrose infusion.
In patient 2, ventricular enlargement was recognized at 9 months of pregnancy. Ultrasonography and MRI demonstrate brain dysgenesis in fetuses with PDHAD.15 Brain function depends on the energy produced from aerobic glucose oxidation. Thus, PDHAD negatively affects brain development. PDHAD should be considered as a differential diagnosis when abnormal brain structure is detected in fetuses. Moreover, when PDHAD is definitely diagnosed, we consider that it is important to administer a ketone formula containing low carbohydrate content and sufficient vitamin B1. Vitamin C, Vitamin E, biotin, coenzyme Q10 and carnitine might also help mitochondrial function because the two patients in this case report had a clinical course without significant change when receiving these supplements; however, the use of these supplements for PDHAD continues to be controversial.16,17,18,19
In conclusion, early and long-term interventions should be tailored according to each patient’s symptoms and problems. Good nutritional conditions should be maintained, while controlling convulsions and preventing aspiration pneumonia.
References
References
Robinson BH, MacMillan H, Petrova-Benedict R, Sherwood WG . Variable clinical presentation in patients with defective E1 component of pyruvate dehydrogenase complex. J Pediatr 1987; 111: 525–533.
Cameron JM, Levandovskiy V, Mackay N, Tein I, Robinson BH . Deficiency of pyruvate dehydrogenase caused by novel and known mutations in the E1alpha subunit. Am J Med Genet A 2004; 131: 59–66.
Kitano A, Endo F, Matsuda I . Immunochemical analysis of pyruvate dehydrogenase complex in 2 boys with primary lactic acidemia. Neurology 1990; 40: 1312–1314.
Awata H, Endo F, Tanoue A, Kitano A, Matsuda I . Characterization of a point mutation in the pyruvate dehydrogenase E1α gene from two boys with primary lactic acidemia. J Inherit Metab Dis 1994; 17: 189–195.
Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW . The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab 2012; 106: 385–394.
Lissens W, De Meirleir L, Seneca S, Liebaers I, Brown GK, Brown RM et al. Mutations in the X-linked pyruvate dehydrogenase (E1) alpha subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat 2000; 15: 209–219.
Bachmann-Gagescu R, Merritt JL 2nd . & Hahn SH . A cognitively normal PDH-deficient 18-year-old man carrying the R263G mutation in the PDHA1 gene. J Inherit Metab Dis 2009; 32 (Suppl 1): 123–126.
Chun K, MacKay N, Petrova-Benedict R, Federico A, Fois A, Cole DE et al. Mutations in the X-linked E1 alpha subunit of pyruvate dehydrogenase: exon skip**, insertion of duplicate sequence, and missense mutations leading to the deficiency of the pyruvate dehydrogenase complex. Am J Hum Genet 1995; 56: 558–569.
Barnerias C, Saudubray JM, Touati G, De Lonlay P, Dulac O, Ponsot G et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol 2010; 52: e1–e9.
DeBrosse SD, Okajima K, Zhang S, Nakouzi G, Schmotzer CL, Lusk-Kopp M et al. Spectrum of neurological and survival outcomes in pyruvate dehydrogenase complex (PDC) deficiency: lack of correlation with genotype. Mol Genet Metab 2012; 107: 394–402.
De Meirleir L, Lissens W, Denis R, Wayenberg JL, Michotte A, Brucher JM et al. Pyruvate dehydrogenase deficiency: clinical and biochemical diagnosis. Pediatr Neurol 1993; 9: 216–220.
Bogaert YE, Sheu KF, Hof PR, Brown AM, Blass JP, Rosenthal RE et al. Neuronal subclass-selective loss of pyruvate dehydrogenase immunoreactivity following canine cardiac arrest and resuscitation. Exp Neurol 2000; 161: 115–126.
Laughton JD, Bittar P, Charnay Y, Pellerin L, Kovari E, Magistretti PJ et al. Metabolic compartmentalization in the human cortex and hippocampus: evidence for a cell- and region-specific localization of lactate dehydrogenase 5 and pyruvate dehydrogenase. BMC Neurosci 2007; 8: 35.
Berendzen K, Theriaque DW, Shuster J, Stacpoole PW . Therapeutic potential of dichloroacetate for pyruvate dehydrogenase complex deficiency. Mitochondrion 2006; 6: 126–135.
Tamaru S, Kikuch,i A, Takagi K, Okuno J, Ishikawa K, Imada S et al. A case of pyruvate dehydrogenase E1α subunit deficiency with antenatal brain dysgenesis demonstrated by prenatal sonography and magnetic resonance imaging. J Clin Ultrasound 2012; 40: 234–238.
Uziel G, Garavaglia B, Di Donato S . Carnitine stimulation of pyruvate dehydrogenase complex (PDHC) in isolated human skeletal muscle mitochondria. Muscle Nerve 1998; 11: 720–724.
Bonne G, Benelli C, De Meirleir L, Lissens W, Chaussain M, Diry M et al. E1 pyruvate dehydrogenase deficiency in a child with motor neuropathy. Pediatr Res 1993; 33: 284–288.
Tischner C, Wenz T . Keep the fire burning: current avenues in the quest of treating mitochondrial disorders. Mitochondrion 2015; 24: 32–49.
Miller MJ, Bostwick BL, Kennedy AD, Donti TR, Sun Q, Sutton VR et al. Chronic oral L-carnitine supplementation drives marked plasma TMAO elevations in patients with organic acidemias despite dietary meat restrictions. JIMD Rep 2016; 30: 39–44.
Wexler ID, Hemalatha SG, McConnell J, Buist NR, Dahl HH, Berry SA et al. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology 1997; 49: 1655–1661.
Wexler ID, Hemalatha SG, Liu TC, Berry SA, Kerr DS, Patel MS . A mutation in the E1 alpha subunit of pyruvate dehydrogenase associated with variable expression of pyruvate dehydrogenase complex deficiency. Pediatr Res 1992; 32: 169–174.
Chun K, MacKay N, Petrova-Benedict R, Robinson BH . Mutations in the X-linked E1 alpha subunit of pyruvate dehydrogenase leading to deficiency of the pyruvate dehydrogenase complex. Hum Mol Genet 1993; 2: 449–454.
Briones P, López MJ, De Meirleir L, Ribes A, Rodés M, Martinez-Costa C et al. Leigh syndrome due to pyruvate dehydrogenase E1 alpha deficiency (point mutation R263G) in a Spanish boy. J Inherit Metab Dis 1996; 19: 795–796.
Lissens W, De Meirleir L, Seneca S, Benelli C, Marsac C, Poll-The BT et al. Mutation analysis of the pyruvate dehydrogenase E1 alpha gene in eight patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat 1996; 7: 46–51.
Marsac C, Benelli C, Desguerre I, Diry M, Fouque F, De Meirleir L et al. Biochemical and genetic studies of four patients with pyruvate dehydrogenase E1 alpha deficiency. Hum Genet 1997; 99: 785–792.
Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Osaka H et al. Biochemical and molecular analysis of an X-linked case of Leigh syndrome associated with thiamin-responsive pyruvate dehydrogenase deficiency. J Inherit Metab Dis 1997; 20: 539–548.
Ostergaard E, Moller LB, Kalkanoglu-Sivri HS, Dursun A, Kibaek M, Thelle T et al. Four novel PDHA1 mutations in pyruvate dehydrogenase deficiency. J Inherit Metab Dis 2009; 32 (Suppl 1): S235–S239.
Data Citations
Kido, Jun HGV Database (2017) http://dx.doi.org/10.6084/m9.figshare.hgv.1358
Acknowledgements
We thank the staff at the Department of Pediatrics at Kumamoto University Hospital. We are grateful to Dr Etsuo Naitou for gene analysis of patient 2, and to Dr Keishin Sugawara for constructive criticism of the manuscript. This case report was partially supported by Grants-in-Aid for Pediatric Research from the Ministry of Health, Labor and Welfare; for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology; for Research on Rare and Intractable Diseases, Health, and Labor Sciences; for guidelines and lifetime medical support systems for inborn errors of metabolism found by newborn screening; for the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development; and for research to revise clinical guidelines and to improve quality of clinical managements for inherited metabolic disorders targeted by newborn screening using tandem mass spectrometry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yoshida, T., Kido, J., Mitsubuchi, H. et al. Clinical manifestations in two patients with pyruvate dehydrogenase deficiency and long-term survival. Hum Genome Var 4, 17020 (2017). https://doi.org/10.1038/hgv.2017.20
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.20
- Springer Nature Limited