
Abstract
Hyperhomocysteinemia (HHcy) is a key risk factor in hepatic steatosis. In this study, we applied a metabolomic approach to investigate the changes in the metabolite profile due to HHcy-induced hepatic steatosis and the effects of omega-3 PUFA (polyunsaturated fatty acid) supplementation in mice. HHcy was induced in mice by giving DL-Hcy (1.8 g/L) in drinking water for 6 weeks, then the mice were sacrificed, and the metabolic profiles of the liver and plasma were analyzed through UPLC-ESI-QTOFMS-based lipidomics. Hepatic triglycerides and cholesterol were further assayed. The expression of ceramide metabolism-related genes was measured by quantitative PCR. Compared with control mice, HHcy mice exhibited hepatic steatosis with a notable increase in ceramide-related metabolites and subsequent upregulation of ceramide synthesis genes such as Sptlc3, Degs2, Cer4 and Smpd4. Omega-3 PUFA was simultaneously administered in HHcy mice through chow diet containing 3.3% omega-3 PUFA supplement for 6 weeks, which significantly ameliorated Hcy-induced hepatic steatosis. The decrease in hepatic lipid accumulation was mainly due to reduced hepatic levels of ceramides, which was partly the result of the lower expression of ceramide synthesis genes, Sptlc3 and Degs2. Similar beneficial effects of DHA were observed in Hcy-stimulated primary hepatocytes in vitro. In summary, Hcy-induced ceramide elevation in hepatocytes might contribute to the development of hepatic steatosis. Furthermore, downregulation of ceramide levels through omega-3 PUFA supplementation ameliorates hepatic lipid accumulation. Thus, ceramide is a potential therapeutic target for the treatment of hepatic steatosis
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) is a chronic liver disease that may progress from steatosis to nonalcoholic steatohepatitis (NASH)1. NAFLD is one of the most common liver disorders, affecting 17%-33% of the general population in the USA2. A recently recognized risk factor of NAFLD is hyperhomocysteinemia (HHcy), whose hallmarks include a sulfur-containing amino acid derived from metabolism of methionine, and elevated plasma Hcy levels (>10 μmol/L) define HHcy. HHcy is an independent risk factor involved in several metabolic disorders3,4,5,6,7. Recently, plasma Hcy levels have been reported to be positively correlated with the development of hepatic steatosis8. In addition, a high-methionine diet (HMD) promotes hepatic steatosis in mice9,10. Although several studies have suggested that HHcy perturbs lipid metabolism via enhanced CD36 expression9,11, the metabolite profile changes and precise targets for the treatment of HHcy-induced hepatic steatosis remain unknown.
Omega-3 fatty acids belong to the family of polyunsaturated fatty acids, which exert various benefits against metabolic syndromes such as obesity and insulin resistance12,13. Omega-3-mediated improvements in lipogenesis and hepatic lipid metabolism have been reported in numerous studies14,15. Other studies have demonstrated that supplementation with omega-3 impedes the development of NAFLD by suppressing of lipogenic genes such as Srebp1c14,16. However, to date, the lipid metabolic profiles of the liver after supplementation with omega-3 remained unclear. In this study, we determined the mechanism underlying omega-3 PUFA's amelioration of hepatic steatosis induced by HHcy.
We found that ceramides are more abundant in HHcy-induced fatty livers, specifically, hepatic levels of saturated ceramides were significantly increased in HHcy mice compared with control mice, possibly because of the upregulation of ceramide synthases in hepatocytes induced by Hcy. Omega-3 supplementation ameliorated HHcy-induced hepatic steatosis through the inhibition of ceramides in the liver, thus suggesting that ceramide may be a potential therapeutic target for the treatment of patients with hepatic steatosis.
Materials and methods
Reagents
DL-homocysteine (Hcy) and docosahexaenoic acid (DHA) were purchased from Sigma-Aldrich Chemical (St Louis, MO, USA). Ceramides were obtained from Avanti Polar Lipids (Alabaster, AL, USA).
Animals
All procedures involving mice were approved by the Peking University Animal Care and Use Committee. Mice (C57BL/6J background) were housed in an animal facility and given free access to water with or without DL-Hcy (1.8 g/L) for 6 weeks as previously described3,4,6. Each group of mice was fed a chow diet either containing 4% fat by weight or supplemented with approximately 3.3% omega-3 PUFAs comprising DHA/EPA (D/E, 3:1) (33 mg/g; a generous gift from Abbott, USA) as previously described in different mouse models17,18. Body weight was measured every 2 weeks during the experiments.
Histological analysis
Oil red O staining was performed on frozen liver sections through a standard protocol, as previously described9.
Triglyceride and cholesterol content quantification
Total triglycerides and total cholesterol in liver were measured with enzymatic kits from Wako Life Sciences (Richmond, VA, USA) and Thermo Scientific (Waltham, MA, USA) as described previously9.
Cell culture
Murine primary hepatocytes were isolated as previously described9,19,20. Hepatocytes were cultured in growth medium, RPMI-1640 medium supplemented with 10% fetal bovine serum. After a 4-h attachment, cells were treated as indicated in the experiments.
Sample preparation for metabolomics analysis
For hepatic lipidomics analysis, approximately 10 mg of liver were homogenized with 200 μL of H2O and then extracted with 1200 μL of a cold chloroform:methanol (2:1) solution containing 2 μmol/L ceramide (19:0) as an internal standard. The homogenate was vortexed at 4 °C for 20 min then centrifuged for 20 min at 13 000 revolutions per minute. The lower organic phase was transferred to a new tube and evaporated under a vacuum. The residue was suspended in 200 μL of a chloroform:methanol (1:1) solution and then diluted with an isopropanol:acetonitrile:H2O (2:1:1) solution.
Lipidomics determination
Samples were analyzed with a Thermo Scientific™ Q Exactive™ hybrid quadrupole Orbitrap mass spectrometer equipped with a Thermo Scientific™ Dionex™ UltiMate™ 3000 Rapid Separation LC (RSLC) system to perform UPLC separations. The UPLC conditions were set as follows. The column was an HSS T3 column (2.1 mm×100 mm, 1.7 μm, Waters) operated at 45 °C. The two UPLC buffers were A- water and B- methanol, both of which contained 0.1% formic acid. The mobile phase gradient was set as: 95% A at 0.1 min; 80% A at 3 min; 25% A at 4.5 min; 0% A at 6.5 min; 0% A at 15 min; 95% A at 15.5 min; 95% A at 17 min. Ceramides and other lipid metabolites were identified by comparing the parent ion mass and MS/MS fragmentations to acknowledged database such as https://metlin.scripps.edu/ or http://www.hmdb.ca. Peak extraction and integration were operated with Xcalibur 2.2 SP1.48 software (Thermo Fisher Scientific, USA), the relative quantification of each metabolite was calculated by comparison to internal standard ceramide (19:0). The mass spectrometer parameters of the full mass scan were set as follows. The pos HESI-II spray voltages were 3.7 kV. The heated capillary temperature was 320 °C, and the heated vaporizer temperature was 300 °C. The sheath gas pressure was 30 psi, and the auxiliary gas pressure was 10 psi. The resolution was 70 000. The auto gain control target was under 1×106. The maximum isolation time was 50 ms, and the m/z range was 150–1500.
Data processing and multivariate data analysis
Xcalibur 2.2 SP1.48 software (Thermo Fisher Scientific, USA) was used for peak extraction and integration. Statistical models, including PLS-DA analysis and heat maps, were created with the MetaboAnalyst 3.0 web service (http://www.metaboanalyst.ca/). The relative level of each analyte was calculated after normalization of the peak area to that of the internal standard.
Statistical analysis
The data are expressed as the mean±SEM. Statistical analyses were performed with two-tailed Student's t-tests and one-way ANOVA with Tukey's confirmation. P values less than 0.05 were considered statistically significant.
Results
HHcy induces hepatic steatosis in vivo
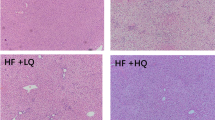
To evaluate the role of HHcy in hepatic steatosis, wild-type C57BL/6J mice were administered drinking water with or without Hcy (1.8 g/L) for 6 weeks. The food consumption, body weight and fasting plasma glucose levels remained unchanged between the HHcy and control mice (Supplementary Figure S1A-S1C). However, Oil red O staining of the liver sections revealed a significant increase in the number of hepatic lipid droplets in the HHcy mice (Figure 1A), which was concomitant with elevated liver weight (Figure 1B) and liver/body mass ratios (Figure 1C). Hepatic triglycerides (Figure 1D) and cholesterol (Figure 1E) were also increased in mice treated with HHcy compared with control. In addition, fatty acid synthesis-related genes, such as fatty acid synthase (Fasn), acetyl-CoA carboxylase (Acc), sterol response element–binding protein 1c (Srebp1c) and peroxisome proliferator-activated receptor gamma (Pparγ) in the liver as well as fatty acid uptake-related genes, including cluster of differentiation 36 (Cd36), were markedly upregulated in HHcy mice. In contrast, the expression of genes involved in fatty acid β-oxidation and TG secretion was not affected in the HHcy treatment group (Figure 1F). These results indicate that HHcy may induce lipid perturbation and hepatic steatosis.

Effects of HHcy on lipid accumulation in vivo. (A) ORO staining of lipids in representative liver sections. (B) Liver weight. (C) Ratios of liver weight/body weight. (D) Hepatic TG content. (E) Hepatic TC content. (F) The mRNA levels of genes involved in hepatic lipogenesis, FFA oxidation, TG secretion and FFA uptake. (A-F) Male C57BL/6 mice (6 weeks old) were treated with vehicle or Hcy (1.8 g/L) for 6 weeks (n=6/group). All data are presented as the mean±SEM. Two-tailed Student's t-test: *P<0.05 compared with vehicle.
The hepatic ceramide levels are significantly elevated after Hcy treatment
To further determine the metabolic profiles of HHcy-induced hepatic steatosis, UPLC-ESI-QTOFMS-based lipidomics analysis was performed to investigate the metabolites in the liver. Heat map analysis and the VIP scores of the biomarkers showed that ceramides were among the metabolites that exhibited differences between the groups (Figure 2A-2C). The hepatic saturated C18:0, C20:0, C22:0 and C24:0 ceramides were significantly upregulated in HHcy mice (Figure 2D), whereas the metabolic profiles of the plasma showed no differences (Supplementary Figure S2A, S2B). These results suggest that HHcy significantly increases liver ceramide levels.

Metabolic profiling of HHcy mice and control mice. (A) Heat map of lipid metabolites in liver. (B) Score scatter plot of the partial least squares discriminant analysis (PLS-DA) model of lipid metabolites in the liver; the red plot represents the vehicle group, and the green plot represents the HHcy group. (C) Variable importance in projection (VIP) plot generated from the PLS-DA, displaying the top 10 most important metabolite features in the liver. (D) The levels of saturated ceramides in the liver. (A-D) Male C57BL/6 mice (6 weeks old) were treated with vehicle or Hcy (1.8 g/L) for 6 weeks (n=4/group). (D) The data are presented as the mean±SEM. Two-tailed Student's t-test: *P<0.05 compared to vehicle.
Hcy promotes ceramide synthesis both in vivo and in vitro
To further investigate the mechanism by which Hcy mediated the increase in ceramide metabolites, ceramide metabolism-related genes in the liver were examined. qPCR analysis showed that the mRNAs encoded by ceramide synthesis-related genes such as serine palmitoyl transferase long chain base 3 (Sptlc3), ceramide synthase 4 (Cers4), degenerative spermatocyte homolog 2 (Degs2) and sphingomyelin phosphodiesterase 4 (Smpd4) were significantly increased in vivo (Figure 3A). Moreover, we found that Hcy also promoted the gene expression of Sptlc3 and Degs2 in primary hepatocytes stimulated with Hcy (100 μmol/L) for 24 h (Figure 3C). However, the mRNAs encoded by genes involved in ceramide catabolism, including sphingomyelin synthase 1 and 2 (Sgms1 and Sgms2), alkaline ceramidase 1 and 2 (Acer1 and Acer2), ceramide kinase (Cerk) and sphingosine kinase1 and 2 (Sphk1 and Sphk2) showed no significant changes compared with corresponding controls in both the livers from Hcy-treated mice and primary hepatocytes treated with Hcy (100 μmol/L) for 24 h (Figure 3B, 3D). Together, these results suggest that Hcy stimulates ceramide synthesis primarily by inducing the expression of genes involved in ceramide synthases in the hepatocytes of mice.

Hcy induces ceramide synthases production both in vivo and in vitro. (A) The mRNA levels of ceramide synthases in the liver. (B) The mRNA levels of ceramide hydrolases in the liver. (A, B) Male C57BL/6 mice (6 weeks old) were treated with vehicle or Hcy (1.8 g/L) for 6 weeks (n=6/group). (C) The mRNA levels of ceramide synthases in primary hepatocytes. (D) The mRNA levels of ceramide hydrolases in primary hepatocytes. (C, D) Primary hepatocytes were treated with Hcy (100 μmol/L) for 24 h (n=5). All data are presented as the mean±SEM. Two-tailed Student's t-test: *P<0.05 compared with vehicle or control as appropriate.
Omega-3 PUFA treatment ameliorates HHcy-induced hepatic steatosis
It has been demonstrated that dietary omega-3 PUFA ameliorates the development of liver dysfunction and steatosis14,21. Therefore, we determined whether HHcy-induced hepatic steatosis might be alleviated by omega-3 PUFA supplementation. Omega-3 PUFA decreased HHcy-induced lipid accumulation in the liver without affecting the food consumption, body weight and fasting plasma glucose levels (Figure 4A, Supplementary Figure S1D–S1F). In line with this, the liver weight and liver/body mass ratios were reduced in mice treated with omega-3 PUFA (Figure 4B, 4C). Moreover, omega-3 PUFA protected from HHcy-augmented hepatic TG and TC accumulation (Figure 4D, 4E) as well as mRNA expression of Fasn, Srebp1c and Pparγ (Figure 4F). These data suggest that omega-3 PUFA might ameliorate HHcy-induced hepatic steatosis.

Administration of omega-3 PUFA attenuates the development of HHcy-induced hepatic steatosis. (A) ORO staining of lipids in representative liver sections. (B) Liver weight. (C) Ratios of liver weight/body weight. (D) Hepatic TG content. (E) Hepatic TC content. (F) The mRNA levels of genes involved in hepatic lipogenesis, FFA oxidation, TG secretion and FFA uptake. Hcy-treated mice were fed a standard chow diet or a chow diet supplemented with omega-3 PUFA (33 mg/g) for 6 weeks and were compared with mice administered vehicle for 6 weeks (n=6/group). All data are presented as the mean±SEM. (B-F) One-way ANOVA with Tukey's correction: *P<0.05 compared with vehicle; #P<0.05 compared to HHcy.
Omega-3 PUFA modifies ceramide metabolism
To determine the changes in the metabolic profiles in the liver after treatment with omega-3 PUFA, UPLC-ESI-QTOFMS-based metabolomics analysis was used to detect the lipid composition. Heat map analysis and a PCA model of the UPLC-ESI-QTOFMS negative mode data from mouse livers showed distinct metabolic profiles among the vehicle-, HHcy- and HHcy+omega3-treated groups (Figure 5A, 5B). Further analysis indicated that omega-3 PUFA abrogated HHcy-induced increases in the saturated ceramide levels in the liver (Figure 5C). These results suggest that the inhibition of ceramides by omega-3 PUFA might lead to improvements in HHcy-induced hepatic steatosis.

Omega-3 PUFA treatment ameliorates the levels of ceramides in the livers of HHcy mice. (A) Heat map of lipid metabolites in the liver. (B) Score scatter plot of the Principal Component Analysis (PCA) model of lipid metabolites in the liver; the red plot represents the vehicle group, the green plot represents the HHcy group and the purple represents the HHcy plus omega-3 group. (C) The saturated ceramide levels in the liver. Hcy-treated mice were fed either a standard chow diet or a chow diet supplemented with omega-3 PUFA (33 mg/g) for 6 weeks and were compared with mice administered vehicle 6 weeks (n=4/group). All data are presented as the means±SEM. One-way ANOVA with Tukey's correction: *P<0.05 compared with vehicle; #P<0.05 compared with HHcy.
Omega-3 PUFA decreases ceramide synthase both in vivo and in vitro
To further investigate the mechanism by which omega-3 PUFA decreased the levels of ceramide metabolites, ceramide metabolism-related genes were examined. qPCR analysis showed that the expression of genes corresponding to ceramide synthase, Sptlc3 and Degs2, were increased by HHcy, and this effect was reversed by omega-3 PUFA supplementation (Figure 6A, 6B). Docosahexaenoic acid (22:6 omega-3, DHA) is a major polyunsaturated fatty acid (PUFA) in the omega-3 series. When pretreatment of 24 h Hcy-stimulated primary hepatocytes with DHA (20 μmol/L), the expression of ceramide synthases were significantly attenuated by DHA (Figure 6C, 6D). Together, these findings indicate that the beneficial effects of omega-3 PUFA on HHcy-induced hepatic steatosis are, at least in part, due to the suppression of ceramide synthases in HHcy mice.

Omega-3 PUFA decreases the expression of ceramide synthases induced by Hcy in hepatocytes. (A) The mRNA levels of ceramide synthases in the liver. (B) The mRNA levels of ceramide hydrolases in the liver. Hcy-treated mice were fed either a standard chow diet or a chow diet supplemented with omega-3 PUFA (33 mg/g) for 6 weeks and were compared with mice administered vehicle for 6 weeks (n=6/group). (C) The mRNA levels of ceramide synthases in primary hepatocytes. (D) The mRNA levels of ceramide hydrolases in primary hepatocytes. Primary hepatocytes were pretreated with DHA (20 μmol/L) for 1 h before Hcy administration (100 μmol/L) for 24 h (n=5). (A-D) All data are presented as the mean±SEM. (A-D) One-way ANOVA with Tukey's correction: *P<0.05 compared with vehicle or control as appropriate; #P<0.05 compared to HHcy or Hcy as appropriate.
Discussion
NAFLD is currently the most common hepatic disorder worldwide22,23,24. Increasing evidence indicates that HHcy is a new risk factor for hepatic steatosis8,9,10. Several studies have reported that HHcy induces hepatic steatosis by disrupting lipid metabolism9,25. Here, we adopted a metabolomic approach to explore the metabolic profiles of hepatic steatosis induced by HHcy and to search for the potential therapeutic targets for treating hepatic steatosis. We found that ceramides (particularly saturated ceramides) were elevated with the development of HHcy-induced hepatic steatosis. Furthermore, the hepatic ceramide levels in HHcy mice supplemented with omega-3 PUFA were significantly decreased, and these mice showed improvements in hepatic lipid accumulation. Interestingly, analysis of the expression of genes involved in ceramide metabolism showed that the HHcy-induced increased ceramide metabolic enzymes in hepatocytes were markedly attenuated in the presence of omega-3 PUFA both in vivo and in vitro.
Ceramide, which serves as the backbone for sphingolipids, has been linked with insulin resistance and NAFLD and is considered as a biomarker for these diseases19,26. A series of studies have revealed that increased ceramides in the liver and plasma are positively correlated with the development of hepatic insulin resistance and steatosis in rodents27,28,29. A clinical study has further confirmed that the concentration of hepatic ceramides is higher in patients with NAFLD26. Furthermore, it has been reported that saturated ceramides such as C16:0 ceramide induce lipid accumulation by upregulating the genes involved in hepatic lipogenesis in a mouse model30. Besides C16:0 Cer, numerous studies have demonstrated that the levels of C18:0, C20:0 and C24:0 saturated ceramides were also increased in HFD-induced hepatic steatosis29,31,32,33,34. The inhibition of the de novo synthesis of saturated ceramides significantly decreased hepatic lipid accumulation and insulin resistance30,34. In line with the results of these studies, we found that ceramides, especially saturated ceramides such as C18:0, C20:0, C22:0 and C24:0, were positively correlated with the progression of HHcy-induced hepatic steatosis. Furthermore, the metabolic profiles in the plasma were unchanged, thus suggesting that the accumulation of ceramides was primarily localized to the liver. Of note, the disruption of hepatic ceramides by omega-3 PUFA led to a decrease in HHcy-induced hepatic steatosis. These results suggested that ceramides play a crucial role in HHcy-induced hepatic steatosis.
In addition, other studies have reported that the inhibition of hepatic ceramide synthesis also ameliorates HFD-induced hepatic steatosis in a mouse model30,35. Collectively, these reports have suggested that inhibiting hepatic ceramide generation and accumulation might be a common target for treating NAFLD and hepatic steatosis.
To explore the mechanism by which Hcy increased the hepatic ceramide levels, we measured the different enzymatic pathways involved in Hcy–regulated ceramide metabolism. Previous studies have revealed that ceramide synthases are responsible for the increase in ceramides36. It has been reported that the genes involved in ceramide synthesis are stimulated by various endogenous factors such as thyroid hormones and angiotensin II37,38. Therefore, ceramide synthase may mediate the actions of these agents or factors as an important second messenger. In the present study, we found that genes involved in ceramide synthesis, such as Sptlc3 and Degs2, were dramatically increased after treatment with Hcy both in vivo and in vitro; additionally, these increases were accompanied by elevated levels of ceramides in HHcy mice. However, abolishing the expression of Sptlc3 and Degs2 by omega-3 significantly ameliorated HHcy-mediated increases of hepatic ceramide, thus suggesting that ceramide synthases were involved in Hcy-induced ceramide production.
Hcy treatment has been reported to significantly promote de novo ceramide synthases in the kidney, thereby potentially contributing to glomerulosclerosis39,40. Further disruption of acid sphingomyelinase, a ceramide-producing enzyme, dramatically attenuates the production of ceramides and improves glomerular oxidative stress induced by HHcy41. In addition, ceramide hydrolytic enzymes (Acer, Sphk, Sgms, Cerk) in the kidney were unchanged after treatment with Hcy, thus further supporting our findings that increased ceramide levels in the livers of HHcy mice were mediated primarily through the ceramide synthases without affecting ceramide hydrolases.
However, it should be noted that this study determined only the levels of ceramide synthase (Sptlc3 and Degs2) genes, and the roles of Sptlc3 and Degs2 in HHcy-induced hepatic steatosis require further elucidation in genetic model animals.
The mechanism of omega-3 PUFA has been demonstrated in different physiological systems, including the regulation of hepatic lipid metabolism42. Omega-3 PUFA supplementation has been reported to ameliorate HFD-induced hepatic steatosis by inhibiting genes involved in lipogenesis, including sterol regulatory element-binding protein 1c (SREBP-1c)14. In addition, dietary omega-3 fatty acids can rescue the fructose-provoked ER stress response, thereby decreasing FFA oxidation and the deposition of hepatic lipids43. In the present study, we showed that omega-3 PUFA ameliorates hepatic steatosis induced by HHcy, possibly as a result of decreases in hepatic ceramide levels and ceramide synthases production. Furthermore, the assembly of DHA into phospholipids in caveolae has been found to lead to decreased ceramide generation and to consequently inhibit cytokine signaling in human retinal endothelial cells44.
In summary, our current findings demonstrate that HHcy-induced ceramide production is involved in the development of hepatic steatosis and that this activity is primarily due to the upregulation of ceramide synthases in hepatocytes. Dietary omega-3 PUFA might abolish HHcy-induced lipid accumulation by decreasing the generation of hepatic ceramides and may serve as a potential therapy for treating patients with hepatic steatosis.
Author contribution
Yong-qiang DONG, **ng-zhong ZHANG, Song-yang ZHANG, Lu-lu SUN, and Hui-ying LIU designed and performed the experiments and analyzed the data; Bo LIU contributed to the histology experiments; **an WANG and Chang-tao JIANG designed and supervised the research; Yong-qiang DONG, Song-yang ZHANG, **an WANG and Chang-tao JIANG wrote and edited the manuscript. All the authors approved the final manuscript.
References
Pagadala M, Kasumov T, McCullough AJ, Zein NN, Kirwan JP . Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol Metab 2012; 23: 365–71.
McCullough AJ . Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol 2006; 40: S17–29.
Li Y, Zhang H, Jiang C, Xu M, Pang Y, Feng J, et al. Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue. J Biol Chem 2013; 288: 9583–92.
Pang Y, Li Y, Lv Y, Sun L, Zhang S, Li Y, et al. Intermedin restores hyperhomocysteinemia-induced macrophage polarization and improves insulin resistance in mice. J Biol Chem 2016; 291: 12336–45.
Sun W, Pang Y, Liu Z, Sun L, Liu B, Xu M, et al. Macrophage inflammasome mediates hyperhomocysteinemia-aggravated abdominal aortic aneurysm. J Mol Cell Cardiol 2015; 81: 96–106.
Li Y, Jiang C, Xu G, Wang N, Zhu Y, Tang C, et al. Homocysteine upregulates resistin production from adipocytes in vivo and in vitro. Diabetes 2008; 57: 817–27.
Zeng X, Dai J, Remick DG, Wang X . Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ Res 2003; 93: 311–20.
Gulsen M, Yesilova Z, Bagci S, Uygun A, Ozcan A, Ercin CN, et al. Elevated plasma homocysteine concentrations as a predictor of steatohepatitis in patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2005; 20: 1448–55.
Yao L, Wang C, Zhang X, Peng L, Liu W, Zhang X, et al. Hyperhomocysteinemia activates the aryl hydrocarbon receptor/CD36 pathway to promote hepatic steatosis in mice. Hepatology 2016; 64: 92–105.
Woo CW, Siow YL, Pierce GN, Choy PC, Minuk GY, Mymin D, et al. Hyperhomocysteinemia induces hepatic cholesterol biosynthesis and lipid accumulation via activation of transcription factors. Am J Physiol Endocrinol Metab 2005; 288: E1002–10.
Morihara N, Ide N, Weiss N . Aged garlic extract inhibits homocysteine-induced scavenger receptor CD36 expression and oxidized low-density lipoprotein cholesterol uptake in human macrophages in vitro. J Ethnopharmacol 2011; 134: 711–6.
Ulven T, Christiansen E . Dietary fatty acids and their potential for controlling metabolic diseases through activation of FFA4/GPR120. Annu Rev Nutr 2015; 35: 239–63.
Huang Q, Wang T, Wang HY . Ginsenoside Rb2 enhances the anti-inflammatory effect of omega-3 fatty acid in LPS-stimulated RAW264.7 macrophages by upregulating GPR120 expression. Acta Pharmacol Sin 2017; 38: 192–200.
Sekiya M, Yahagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, et al. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 2003; 38: 1529–39.
Dossi CG, Tapia GS, Espinosa A, Videla LA, D'Espessailles A . Reversal of high-fat diet-induced hepatic steatosis by n-3 LCPUFA: role of PPAR-alpha and SREBP-1c. J Nutr Biochem 2014; 25: 977–84.
Raptis DA, Limani P, Jang JH, Ungethum U, Tschuor C, Graf R, et al. GPR120 on Kupffer cells mediates hepatoprotective effects of omega3-fatty acids. J Hepatol 2014; 60: 625–32.
Xu P, Wang H, Kayoumu A, Wang M, Huang W, Liu G . Diet rich in Docosahexaenoic Acid/Eicosapentaenoic Acid robustly ameliorates hepatic steatosis and insulin resistance in seipin deficient lipodystrophy mice. Nutr Metab (Lond) 2015; 12: 58.
Zhang X, Yang N, Ai D, Zhu Y . Systematic metabolomic analysis of eicosanoids after Omega-3 polyunsaturated fatty acid supplementation by a highly specific liquid chromatography-tandem mass spectrometry-based method. J Proteome Res 2015; 14: 1843–53.
Jiang C, **e C, Lv Y, Li J, Krausz KW, Shi J, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 2015; 6: 10166.
Zhou B, Zhou DL, Wei XH, Zhong RY, Xu J, Sun L . Astragaloside IV attenuates free fatty acid-induced ER stress and lipid accumulation in hepatocytes via AMPK activation. Acta Pharmacol Sin 2017; 38: 998–1008.
Soni NK, Ross AB, Scheers N, Savolainen OI, Nookaew I, Gabrielsson BG, et al. Eicosapentaenoic and docosahexaenoic acid-enriched high fat diet delays skeletal muscle degradation in mice. Nutrients 2016; 8. pii: E543.
Angulo P . Nonalcoholic fatty liver disease. New Engl J Med 2002; 346: 1221–31.
Fan JG, Zhu J, Li XJ, Chen L, Li L, Dai F, et al. Prevalence of and risk factors for fatty liver in a general population of Shanghai, China. J Hepatol 2005; 43: 508–14.
Liu MX, Gao M, Li CZ, Yu CZ, Yan H, Peng C, et al. Dicer1/miR-29/HMGCR axis contributes to hepatic free cholesterol accumulation in mouse non-alcoholic steatohepatitis. Acta Pharmacol Sin 2017; 38: 660–71.
Robert K, Nehme J, Bourdon E, Pivert G, Friguet B, Delcayre C, et al. Cystathionine beta synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterology 2005; 128: 1405–15.
Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol 2016; 64: 1167–75.
Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 2007; 5: 167–79.
Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 2014; 20: 687–95.
Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 2014; 20: 678–86.
Jiang C, **e C, Li F, Zhang L, Nichols RG, Krausz KW, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 2015; 125: 386–402.
Yang L, ** GH, Zhou JY . The role of ceramide in the pathogenesis of alcoholic liver disease. Alcohol Alcohol 2016; 51: 251–7.
Liangpunsakul S, Sozio MS, Shin E, Zhao ZW, Xu Y, Ross RA, et al. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am J Physiol Gastrointest Liver Physiol 2010; 298: G1004–12.
Bergman BC, Brozinick JT, Strauss A, Bacon S, Kerege A, Bui HH, et al. Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia 2016; 59: 785–98.
Cinar R, Godlewski G, Liu J, Tam J, Jourdan T, Mukhopadhyay B, et al. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology 2014; 59: 143–53.
Kurek K, Piotrowska DM, Wiesiolek-Kurek P, Lukaszuk B, Chabowski A, Gorski J, et al. Inhibition of ceramide de novo synthesis reduces liver lipid accumulation in rats with nonalcoholic fatty liver disease. Liver Int 2014; 34: 1074–83.
Boslem E, Meikle PJ, Biden TJ . Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012; 4: 177–87.
Lehtonen JY, Horiuchi M, Daviet L, Akishita M, Dzau VJ . Activation of the de novo biosynthesis of sphingolipids mediates angiotensin II type 2 receptor-induced apoptosis. J Biol Chem 1999; 274: 16901–6.
Dbaibo GS, El-Assaad W, Krikorian A, Liu B, Diab K, Idriss NZ, et al. Ceramide generation by two distinct pathways in tumor necrosis factor alpha-induced cell death. FEBS Lett 2001; 503: 7–12.
Yi F, Zhang AY, Li N, Muh RW, Fillet M, Renert AF, et al. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney Int 2006; 70: 88–96.
Yi F, Zhang AY, Janscha JL, Li PL, Zou AP . Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney Int 2004; 66: 1977–87.
Boini KM, **a M, Li C, Zhang C, Payne LP, Abais JM, et al. Acid sphingomyelinase gene deficiency ameliorates the hyperhomocysteinemia-induced glomerular injury in mice. Am J Pathol 2011; 179: 2210–9.
Scorletti E, Byrne CD . Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu Rev Nutr 2013; 33: 231–48.
Zheng JY, Peng C, Ai YB, Wang H, **ao XQ, Li JB . Docosahexaenoic acid ameliorates fructose-induced hepatic steatosis involving ER stress response in primary mouse hepatocytes. Nutrients 2016; 8. pii: E55.
Opreanu M, Lydic TA, Reid GE, McSorley KM, Esselman WJ, Busik JV . Inhibition of cytokine signaling in human retinal endothelial cells through downregulation of sphingomyelinases by docosahexaenoic acid. Invest Ophthalmol Vis Sci 2010; 51: 3253–63.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 91439206, 31230035, 81470554, and 81522007), the National Key Research and Development Program of China (2016YFC0903100) and the 111 Project of the Chinese Ministry of Education (No B07001).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available on the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Figures
Supplementary Figure S1–S2 (DOC 2193 kb)
Rights and permissions
About this article

Cite this article
Dong, Yq., Zhang, Xz., Sun, Ll. et al. Omega-3 PUFA ameliorates hyperhomocysteinemia-induced hepatic steatosis in mice by inhibiting hepatic ceramide synthesis. Acta Pharmacol Sin 38, 1601–1610 (2017). https://doi.org/10.1038/aps.2017.127
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.127
- Springer Nature Singapore Pte Ltd.
Keywords
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.