
Abstract
Genome modification platforms are fast becoming valuable tools for the development of novel therapies. The use of sequence-specific DNA binding proteins in conjunction with various effector domains enables targeted gene editing to be employed as a mode of therapy. Although this field of research has largely focused on the engineering and repair of mammalian genes, the technology may also be used to disrupt viral DNA or host factors associated with pathogenesis of viral disease. For persistent or latent infections, targeted mutagenesis of the episomal or proviral DNA could render the virus inactive. Alternatively, virus-resistant cells may be generated by disrupting the expression of host factors that are required for viral infection. This review highlights some of the approaches currently used to disable viruses, with a particular focus on TALENs and repressor TALEs for antiviral therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Several viruses that replicate using DNA intermediates are significant causes of global mortality and morbidity. These include human immunodeficiency virus-1 (HIV-1), hepatitis B virus (HBV), herpes simplex virus-1 (HSV-1), human T cell lymphotropic virus (HTLV), and human papilloma virus (HPV). The viral DNA templates are capable of reactivating replication and serve as latent reservoirs for the virus. Since available therapies do not affect viral DNA directly, therapeutic inactivation of these sequences by targeted gene editing would be a powerful means of disabling viral replication permanently. Developments in engineering customized DNA-binding proteins have therefore been of interest to advancing gene therapy for the treatment of several chronic viral infections. The main classes of sequence-specific binding proteins that are being developed for therapeutic application are:
-
1.
Transcription activator-like effectors (TALEs),
-
2.
Zinc finger proteins (ZFPs),
-
3.
Clustered regularly interspaced palindromic repeats (CRISPR) with CRISPR-associated nuclease (Cas) systems, and
-
4.
Homing endonucleases (HEs) or meganucleases.
The CRISPR/Cas system differs from the others in that base pairing of a guide RNA determines specificity of the gene editors. Interaction of TALEs, ZFPs, and HEs with their targets is determined by protein binding to the DNA target. Specificity, affinity of binding to intended targets and efficiency with which therapeutic functions may be engineered is influenced by the class of gene editor and the targets to which they bind. A comparison of the properties of the currently available engineered sequence-specific DNA binding proteins is provided in Table 1. Derivatives of TALEs have compared favorably to other DNA binding proteins and have exciting potential for antiviral gene therapy.
Ensuring specificity of interaction with targets is critically important for clinical use of genome editing and will be a major determinant of therapeutic success of targeted gene editing. To keep off-target interaction to a minimum, it is estimated that at least 17 bases within a viral DNA sequence need to be targeted [1]. The probability of a random 17 bp target site within a viral sequence occurring within the human genome is approximately 0.15. For a target of 18 bp, the probability is roughly 0.02. Although this should enable the design of elements that target viral DNA with good specificity, inherent incomplete specificity for targets and presence of human sequences that have homology to viral targets influence the precision of gene editing. To avoid toxicity, targets that have partial homology to host exonic or transcriptional regulatory elements should also be avoided. In addition to targeting viral sequences, disabling host factors that are required for viral infection is another potential antiviral therapeutic application of gene editing. Data indicate that this approach may be used to disrupt the CCR5 co-receptor in autologous CD4 T cells and have potential for HIV-1 therapy [2•].
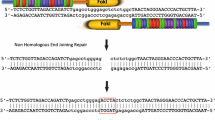
Formation of double-stranded breaks (DSBs) at specific sites of viral DNA is the basis for gene editing with customized nucleases. Normally, such DSBs are repaired by cellular machinery that is responsible for error-prone non-homologous end joining (NHEJ). In most cases, ligation of the cleaved DNA restores the sequence, but insertions and deletions (indels) may occur following repeated target cleavage. This disruption of a viral sequence at the target site could disable replication. Homology-directed repair (HDR) may also be harnessed by introducing sequences with homology to DNA flanking the cleavage target at the same time as cleavage by a sequence-specific nuclease. HDR is more efficient following the formation of DSBs. This property has been exploited to achieve targeted insertion and genome modification (recently reviewed in [3–6]). In addition to the use for therapy, targeted genome editing has been employed to generate model organisms, such as ‘knock-in’ murine lines.
To inhibit gene expression in a targeted way, the Krüppel-associated box (KRAB) has been fused to DNA-targeting proteins [7]. The KRAB domain is naturally found in transcriptional regulatory proteins [8], and the potent inhibitory effect is mediated by recruitment of KRAB-associated protein 1 (KAP1), which in turn engages the function of other factors such as heterochromatin protein 1 (HP1) and various histone-modifying proteins [9]. The formation of heterochromatin causes potent transcriptional suppression [10]. This effect may be sustained and is effective over long ranges of DNA, which is potentially useful to counter viral replication.
Use of TALEs for Genome Modification
Transcription activator-like effectors (TALEs) are naturally found in species of the Xanthomonas genus of pathogenic plant bacteria. They infect a wide variety of plants, and pathogenic forms may be destructive of agricultural crops [11]. Naturally, TALEs function by regulating the host cells’ gene expression to enhance survival of the bacteria. To achieve this, TALEs are introduced into the host plant cells via needle-like structures of a type III secretory system [12]. AvrBs3, one of the best characterized TALEs, increases expression of UPA20 [13]. This helix-loop-helix-containing transcriptional factor is a master regulator of plant cell growth and causes a cascade of cellular events that leads to hypertrophy of plant cells.
Natural TALEs are made up of three main domains:
-
1.
A nuclear localization signal (nls) at the C terminal end,
-
2.
An acid activation domain (AD) that is responsible for transcription activation, and
-
3.
A central region that comprises a tandem arrangement of modules and confers sequence-specific DNA binding properties on the TALEs.
Specificity of target gene regulation by TALEs is achieved by interaction of DNA-binding modules with their targets [14, 15]. This property of TALEs attracted interest of researchers working on targeted gene editing. Each module of the DNA-binding domain comprises 33 to 35 amino acids (reviewed in [12, 16, 17]). Typically, 18.5 modular repeats constitute a DNA-binding domain, and each interacts with a single nucleotide in a DNA sequence. The C-terminal module usually comprises of 20 amino acids and is therefore indicated as half a module. With the exception for the amino acids at positions 12 and 13, the sequence of amino acids in each of the modules is similar. The two residues at positions 12 and 13 constitute the repeat-variable diresidues (RVDs) and confer nucleotide-binding specificity on each module.
Deciphering the specificity of RVDs’ nucleotide binding [14, 15] was a significant development and has enabled advancing use of TALE derivatives for antiviral therapy. There is some degeneracy to the code and more than one RVD may bind to the same base. Conversely, the same RVD may bind to different bases. RVDs comprising NN (Asn-Asn), NI (Asn-Ile), NG (Asn-Gly), and HD (His-Asp) have been used to bind to G, A, T, and C, respectively. However, NN may also interact with A residues. NK (Arg-Lys) is more specific for G, but the binding affinity of TALEs with modules containing these RVDs may be lower [18]. NH (Asn-His) seems to have high affinity for G, but does not compromise TALE activity [19]. A significant advantage of using TALEs to bind to specific DNA sequences is that individual monomers may be coupled without the neighboring sequences significantly influencing binding affinity. This is contrary to the case with engineering of ZFPs to bind specific targets. Arranging monomer-encoding sequences to constitute an artificial TALE is therefore convenient to engineer proteins with desired DNA-binding affinity [17].
General Principles of using Gene Editing to Disable Viral Replication
Using genome engineering agents to cleave episomal viral DNA typically harnesses the error-prone NHEJ pathway to introduce indels at specific sites of the viral DNA. This approach to viral gene therapy has recently gained considerable interest as it potentially causes permanent inactivation of viral replication. To date, only a few potential viral targets have been investigated. These include two retroviruses (HIV-1 and HTLV) and three DNA viruses (HSV, HPV, and HBV). All four main classes of sequence-specific binding proteins, linked to various DNA cleavage enzymes, have been investigated as potential antiviral agents (Table 2). Both ZFNs and CRISPR/Cas9 nucleases have been designed to cleave long terminal repeats (LTRs) of the proviral DNA of HIV-1 and HTLV. Resulting reduced viral gene expression and elimination of the integrated proviral DNA have been demonstrated [26–28, 34]. HSV antiviral activity has been observed following site-directed cleavage of the episomal DNA using HEs [31, 32] and CRISPR/Cas9 endonucleases [33] in cell culture models of infection. In addition, Aubert and colleagues used an adeno-associated viral vector (AAV) to deliver these anti-HSV HEs [32]. Success of the approach indicates that the vector platform may have therapeutic utility. Efficient cleavage of HPV was achieved by Mino et al., who generated zinc finger fusion proteins linked to the staphylococcal nuclease domain, which subsequently resulted in a decrease in viral replication in vitro [30]. Finally, HBV gene targeting and viral inactivation have been achieved using ZFNs [20, 21] and TALENs [22•, 23] in cell culture and animal models of infection and/or replication. To date, this TALEN-mediated direct antiviral approach has only been described for HBV.
Anti-HBV TALENs
Chronic HBV infection is associated with persistence of the episomal covalently closed circular DNA (cccDNA) molecules which exist as stable minichromosome-like structures in the nucleus of infected hepatocytes [35, 36]. HBV’s compact genome arrangement limits sequence plasticity, which makes it a good candidate for nuclease-mediated therapy as the likelihood of emergence of viral escape mutants is low. Initial investigations by Cradick and colleagues illustrated that engineered ZFNs were able to cleave HBV DNA targets in cultured cells [20]. By targeting the HBV core (C) open reading frame, they achieved 36 % disruption at the cognate binding site when using a replication-competent plasmid-based cell culture model of viral replication. In 2013, Bloom et al. showed that TALENs were also capable of efficiently cleaving HBV DNA. This resulted in the inactivation of viral replication in both cultured liver-derived cells and also in a murine model of HBV replication [22•]. TALENs designed to target overlap** reading frames within the surface (S) and C regions inhibited expression of proteins encoded by the sequences. The S TALEN-mediated cleavage of up to 35 % of cccDNA copies in the HepG2.2.15 cell line. Further validation of these two TALEN pairs was established in vivo when murine hydrodynamic injection was used to co-deliver an HBV replication competent plasmid together with DNA encoding the anti-HBV TALENs [22•]. Between 58 and 87 %, targeted disruption was observed, without any apparent TALEN-associated liver toxicity. Deep sequencing of the S and C target sites demonstrated that deletions were the predominant mutation. Cleavage of the HBV DNA also reduced the number of circulating viral particles, indicating disruption of HBV replication. Later in 2013, these results were supported by Chen and colleagues [23], who also showed that TALENs designed to bind to the C and polymerase (P) open reading frames reduced viral antigen and pregenomic RNA expression in vitro and in vivo. They found that the antiviral effect was not limited to a single genotype, suggesting that a TALEN pair may be used to target multiple genotypes. Data from this study also showed that TALENs may be used together with IFN-α, a licensed anti-HBV agent, to enhance anti-HBV efficacy in a synergistic manner.
Use of rTALEs for Transcriptional Silencing of HBV Genes
The minichromosome-like structure of HBV cccDNA may also be amenable to transcriptional gene silencing through induction of heterochromatin formation. Feasibility of the approach was demonstrated with the first use of sequence-specific DNA binding proteins to counter HBV replication. Polydactyl zinc finger proteins (ZFPs), engineered by fusing three to six ZFPs as a single array and designed to bind to enhancer regions of duck hepatitis B virus, reduced pregenomic RNA and viral protein expression [37]. The effect was most likely to have been a result of interference with the binding of transcription factors to viral cis elements. The investigation of anti-HBV TALENs demonstrated that a TALEN pair targeting the P open reading frame (P1) was capable of inhibiting viral replication without causing DNA cleavage [22•]. As this P1 TALEN target site overlaps with the HBV enhancer I sequence, it is likely to be acting by a similar mechanism to that of the duck hepatitis B virus ZFPs. To augment an inhibitory effect, the KRAB domain was fused to the P1 TALE to generate a repressor TALE (rTALE). Highly efficient inhibition of viral replication was demonstrated in cultured cells and in vivo (our unpublished data). A recent study by Uhde-Stone and colleagues suggests that the position of the TALE binding domain may be as important as the role of the effector [38••]. Despite the presence of the activation domain, TALE-activators positioned downstream of the promoter region of a reporter cassette resulted in transcriptional repression. Whether this is important for therapeutic gene silencing of viral genes, such as those of HBV, needs to be explored.
Limitations of Gene Editing for HBV Therapy
A major concern of any nuclease-mediated gene therapy is the potential for genotoxic events as a result of off-target cleavage [39–41]. Bloom and colleagues reported bioinformatic analysis showing no potential off-target binding sites in either the mouse or human genomes for TALENs targeting S, C, and P regions of HBV [22•]. This finding may explain the absence of any TALEN-associated cytotoxicity in their study. HBV DNA may therefore be a good target for genome engineering, since the likelihood of genotoxicity appears low. Since hydrodynamic injection for the delivery of TALENs and rTALEs is not clinically useful, hepatotropic vectors are required for therapeutic use. AAV vectors have been used to successfully deliver anti-HBV ZFNs in a cell culture model of HBV infection [21]. However, the large size of TALEN-encoding sequences, together with the requirement of the nucleases to function as dimers, places limitations on the application of AAVs to delivery of the sequences. The size limit of transgenes that may be incorporated into single-stranded AAVs is approximately 4.5 kb. As a result, two vectors are required to deliver sequences that express a complete TALEN dimer, but a single vector may be used to transduce cells with an rTALE.
The repetitive nature of the sequences encoding the TALE DNA binding domain also places a restriction on the type of viral vector that may be employed to deliver these gene editing sequences. This was confirmed in a study by Holkers et al. who demonstrated that TALENs may be efficiently incorporated into recombinant adenoviruses, but use of lentiviruses to deliver the sequences is suboptimal because these vectors are prone to recombination [42]. Although anti-HBV TALENs and rTALEs present a unique approach to viral gene therapy, challenges of ensuring specificity for HBV targets and efficient delivery to hepatocytes in vivo need to be achieved before the technology is clinically applicable.
Engineering Resistant Host Cells
An alternative approach to treating viral infections, which is currently being investigated for hepatitis C virus (HCV) and HIV-1, is to disrupt host genes that are required for active viral infection and/or replication. Since viruses rely on host cell transcription factors, proteins, and enzymes for attachment, replication, and virion assembly, inhibition of host factors may be used to counter viral infections. Moreover, by disabling host factors, risk of viral escape is also minimized. The reason is that the risk of a virus generating a mutation that enables it to bypass an essential cellular stage of its replication is very unlikely.
Gene Editing to Render T Cells Resistant to HIV-1 Infection
The current treatment for HIV-1 involves use of a combination of antiretroviral drugs. The treatment regimen is referred to as highly active antiretroviral therapy (HAART), and its use has dramatically improved patient survival. However, HAART is not able to eradicate the virus from infected individuals and is not curative of HIV-1 infection. Moreover, treatment interruption and/or non-adherence to the therapy may lead to the emergence of drug-resistant viruses [43]. Thus, there is a clear need to develop novel strategies for controlling viral replication. The natural replication cycle of HIV-1 offers a wide range of steps that may be targeted for therapeutic purposes.
An attractive approach aims to block initial stages of HIV-1 infection. The rationale for such a strategy has been provided by experiences with the single success of curing a patient of HIV-1 infection. Timothy Brown, also known as ‘The Berlin Patient’, was HIV-1-positive and underwent hematopoietic stem cell (HSC) transplantation for the treatment of acute myeloid leukemia (AML) [44]. He received the transplant from a donor who harbored a homozygous 32-bp deletion in the C–C chemokine receptor (5CCR5) gene. This Δ32 mutation leads to premature termination of translation with consequent loss of protein expression. Cells lacking CCR5 are naturally resistant to HIV-1 infection since the protein is used as co-receptor for cellular entry by the CCR5-tropic HIV-1 virus. Moreover, HIV-1 disease progression seems to correlate with CCR5 dose, as development of clinical complications of HIV-1 infection proceeds more slowly in individuals who are heterozygous for the Δ32 mutation. This evidence has established cellular CCR5 as a good target for antiviral strategies aims at disrupting infection by preventing interaction of the virus with its co-receptor [45, 46].
Knocking Out CCR5
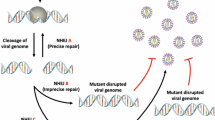
Although potential donors who are homozygous for the Δ32 mutation in CCR5 are rare [47], gene editing ex vivo may be used to knock-down or completely eliminate CCR5 expression in patients’ cells. By using autologous cells, as depicted in Fig. 1, the risks associated with heterologous haematopoietic stem cell (HSC) transplantation and chemotherapy regimens are diminished. Moreover, by eliminating a key element that is required for virus entry, the chance of selecting esca** mutants is minimized.

Viral and host genome modification strategies. TALEN pairs and rTALEs may be used either in an in vivo or ex vivo context. Each TALEN comprises of a DNA binding domain fused to a C-terminal nuclease domain, most commonly FokI. rTALEs comprise of a DNA binding domain fused to an N-terminal repressor domain, such as KRAB. Incorporation of the TALE-based modification agents into viral vectors (adenoviral or AAV packaging systems) or liposomal non-viral vectors facilitates delivery in vivo. Furthermore, tissue-specific vectors may be used to target viral DNA at the site of infection. To generate host cells that are resistant to viral infection, ex vivo genome modification is required. HSCs or CD34+ cells are first isolated from the patient and maintained in a cell culture environment before genome modification. TALEs and rTALEs may be delivered directly to the cells as proteins, or by electroporation of the encoding DNA/RNA. Alternatively, viral and non-viral vectors may be employed. The autologous-modified cells are then selected, enriched, and reinfused into the patient
Tebas et al. have recently shown that HIV-resistant T-Cells lacking the CCR5 receptor could be generated by using CCR5-specific ZFNs. Upon transplantation of HIV-1-infected patients with autologous-modified T cells, the CCR5 mutation caused a lower viral load when HAART treatment was interrupted [2•]. This is a milestone in the field of ex vivo gene editing as it was the first clinical trial to show some benefit in human patients transplanted with engineered autologous T cells. An important observation of the study was that treatment of one of the patients was particularly effective, and only later was he was discovered to be heterozygous for the Δ32 mutation of CCR5. This finding highlights the correlation between CCR5 levels and HIV-1 resistance, and also emphasises the importance of achieving bi-allelic CCR5 knockout.
Although these results are promising, the genotoxic risk associated with off target cleavage by designer nucleases is a concern. Potential problems resulting from off target effects have prevented widespread use, particularly in applications that involve stem cells (i.e., CD34+ cells) with high-proliferative capacity [48]. Indeed, studies have shown that CCR5-specific ZFNs, besides being highly active at their intended target, may cleave at additional genomic sites that share some homology with the target site [49, 50]. Non-specific cleavage is particularly important for targeted disruption of CCR5, since this gene has a paralogue, CCR2, on the same chromosome. This off target site is efficiently cleaved by CCR5-specific ZFNs and increases risk of genomic instability. Recently, in a side-by-side comparison with ZFNs, TALENs were better able to discriminate between very similar target sites of CCR5 and CCR2 in the human genome [29]. TALENs typically recognize longer target sequences compared to ZFNs, and this might be the major determinant for the reduced off target cleavage that was observed. In addition, the use of obligate heterodimeric cleavage domains may further increase the safety associated with the use of TALENs.
Challenges Facing Anti-HIV-1 Gene Editing Carried Out Ex Vivo
Availability of a nuclease platform that causes minimal off target cleavage [51] creates important opportunities for clinical translation of ex vivo targeted genome editing. However, a hindrance to the use of gene editing to treat HIV-1 infection is the difficulty of achieving the short duration of high expression of the gene editors that is required for bi-allelic modification of CCR5 without off target mutation. Delivery methods that entail the use of viral or non-viral vectors have been tested. In the aforementioned clinical trial reported by Tebas and colleagues [2•], ZFNs were delivered using recombinant adenoviral vectors. The high-viral doses that are necessary may be associated with induction of an immune response to the adenoviral vectors and may limit further infusion of cells following ex vivo-based treatments. Direct plasmid transfection causes activation of an innate immune response and may therefore be unsuitable. An alternative method to express the designer nucleases in primary T cells or hematopoietic stem cells may be achieved by electroporation of mRNA encoding for the engineered nucleases. This method combines high efficiency and only minimal cytotoxicity. However, the large amount of mRNA that is needed may pose cost-associated problems for the generation of clinical-grade reagents. Direct protein delivery may be an appropriate alternative. Dose would be easier to control but delivery to the target cells may still be problematic. Since protein purification is time-consuming, associated with high production costs and contaminants may be toxic, clinical use of recombinant gene editing proteins will require further refinement.
Using TALENs to Control HCV Infection and Replication
HCV, a hepatotropic RNA virus, is clinically managed through a genotype-specific combinatorial therapy approach reviewed by [52]. For most patients treatment regimens include peginterferon and ribavirin and a third therapeutic may be added in some cases. A number of adverse side effects and the emergence of viral escape mutants are major drawbacks of currently available therapies. Recently, two independent studies have demonstrated that TALEN-based disruption of host genes in liver derived cells inhibits HCV replication and cell entry [24, 25]. An APOB -/- Huh-7 stable cell line was established to show that the expression of the apoB-100 protein is necessary for active HCV replication [24]. Similarly, cell entry and replication cycle of HCV may be obstructed by using TALENs to knock out the diacylglycerol acyltransferase-1 (DGAT1) gene [25]. Although these are interesting observations, the feasibility of using TALENs for treating HCV infection remains to be established. Exclusion of toxic effects of mutating a natural host protein will be important.
Conclusions
Very impressive progress has been made in develo** antiviral application of customized DNA sequence-specific artificial rTALEs and TALENs. By introducing mutations at intended sites, TALENs are capable of achieving permanent inhibition of viral replication. With rTALEs, the potential for unintended mutation is limited. However, durable inhibition of viral gene expression may be attained. As with most gene therapy strategies, the challenges that face advancing rTALEs and TALENs to a stage of clinical application are significant. Some of the hurdles that need to be overcome, and which are generic to gene therapy, are the following:
-
1.
Ensuring efficient delivery to intended virus-infected target tissues,
-
2.
Controlling the dose of therapeutic rTALEs and TALENs,
-
3.
Ensuring adequate specificity,
-
4.
Limiting host’s immune responses to the rTALEs and TALENs, and
-
5.
Preventing viral escape.
Durable expression may be complicated by increased risk of unintended off target effects. The delivery of mRNA encoding the rTALEs and TALENs or use of recombinant proteins, instead of protein-encoding DNA cassettes, may address this limitation. This should ensure that transient and more regulatable effects are achieved. Ex vivo delivery of rTALEs and TALENs may be easier, but technically demanding procedures for modifying cells in culture and reinfusion into patients limits widespread use. Despite these challenges, the use of rTALEs and TALENs for treatment of viral infections is poised at an exciting stage. Significant developments in their application, together with that of other gene editing technologies, are surely imminent.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Schiffer JT et al. Targeted DNA mutagenesis for the cure of chronic viral infections. J Virol. 2012;86(17):8920–36.
Tebas P et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–10. This study describes the results obtained from the first nuclease-mediated CCR5 knockout clinical trial that was conducted using ZFNs on patients harbouring HIV.
Gaj T, Gersbach CA, Barbas III CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405.
Wei C et al. TALEN or Cas9—rapid, efficient and specific choices for genome modifications. J Genet Genomics. 2013;40(6):281–9.
Li HL, Nakano T, Hotta A. Genetic correction using engineered nucleases for gene therapy applications. Develop Growth Differ. 2014;56(1):63–77.
Sakuma T, Woltjen K. Nuclease-mediated genome editing: at the front-line of functional genomics technology. Develop Growth Differ. 2014;56(1):2–13.
Beerli RR et al. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci U S A. 1998;95(25):14628–33.
Huntley S et al. A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006;16(5):669–77.
Friedman JR et al. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996;10(16):2067–78.
Groner AC et al. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010;6(3):e1000869.
Mew T et al. Focus on bacterial blight of rice. Plant Dis. 1993;77(1):5–12.
Boch J, Bonas U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol. 2010;48:419–36.
Kay S et al. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science. 2007;318(5850):648–51.
Boch J et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–12.
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501.
Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14(1):49–55.
Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng. 2013;110(7):1811–21.
Huang P et al. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29(8):699–700.
Streubel J et al. TAL effector RVD specificities and efficiencies. Nat Biotechnol. 2012;30(7):593–5.
Cradick TJ et al. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol Ther. 2010;18(5):947–54.
Weber ND et al. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS One. 2014;9(5):e97579.
Bloom K et al. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther. 2013;21(10):97–1889. This study demonstrates the activity of TALENs against HBV, including repression of viral transcription. Description of the direct delivery and activity of TALENs is shown in a murine model.
Chen J, et al., An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol Ther. 2013. This study illustrates that antiHBV TALENs can be used in combination with IFN-α to achieve an enhanced therapeutic effect.
Ding Q et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12(2):238–51.
Sung PS et al. Hepatitis C virus entry is impaired by claudin-1 downregulation in diacylglycerol acyltransferase-1-deficient cells. J Virol. 2014;88(16):44–9233.
Qu X et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013;41(16):7771–82.
Ebina H et al. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510.
Hu W et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014;111(31):11461–6.
Mussolino C et al. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014;42(10):6762–73.
Mino T et al. Gene- and protein-delivered zinc finger-staphylococcal nuclease hybrid for inhibition of DNA replication of human papillomavirus. PLoS One. 2013;8(2):e56633.
Grosse S et al. Meganuclease-mediated inhibition of HSV1 infection in cultured cells. Mol Ther. 2011;19(4):694–702.
Aubert M et al. In vitro inactivation of latent HSV by targeted mutagenesis using an HSV-specific homing endonuclease. Mol Ther Nucleic Acids. 2014;3:e146.
Bi Y et al. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 2014;10(5):e1004090.
Tanaka A et al. A novel therapeutic molecule against HTLV-1 infection targeting provirus. Leukemia. 2013;27(8):1621–7.
Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64(1):51–68.
Newbold JE et al. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol. 1995;69(6):3350–7.
Zimmerman KA et al. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J Virol. 2008;82(16):8013–21.
Uhde-Stone C, Cheung E, Lu B. TALE activators regulate gene expression in a position- and strand-dependent manner in mammalian cells. Biochem Biophys Res Commun. 2014;443(4):94–1189. This study demonstrates how the TALE DNA binding domain can affect transcription irrespective of the effector domain, thus emphasizing the importance of selecting an appropriate target site.
Richardson C, Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405(6787):697–700.
Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12(9):440–50.
Bree RT et al. The switch from survival responses to apoptosis after chromosomal breaks. DNA Repair (Amst). 2004;3(8–9):989–95.
Holkers M et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41(5):e63.
Barichievy S et al. The inhibitory efficacy of RNA POL III-expressed long hairpin RNAs targeted to untranslated regions of the HIV-1 5′ long terminal repeat. Oligonucleotides. 2007;17(4):419–31.
Hutter G et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692–8.
de Roda Husman AM et al. Association between CCR5 genotype and the clinical course of HIV-1 infection. Ann Intern Med. 1997;127(10):882–90.
Liu R et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86(3):367–77.
Kiem HP et al. Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell. 2012;10(2):137–47.
Mussolino C, Cathomen T. On target? Tracing zinc-finger-nuclease specificity. Nat Methods. 2011;8(9):725–6.
Gabriel R et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29(9):816–23.
Pattanayak V et al. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8(9):765–70.
Osborn MJ et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21(6):1151–9.
Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med. 2013;368(20):1907–17.
Compliance with Ethics Guidelines
Conflict of Interest
Kristie Bloom, Claudio Mussolino, and Patrick Arbuthnot declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Genome Editing
Rights and permissions
About this article

Cite this article
Bloom, K., Mussolino, C. & Arbuthnot, P. Transcription Activator-Like Effector (TALE) Nucleases and Repressor TALEs for Antiviral Gene Therapy. Curr Stem Cell Rep 1, 1–8 (2015). https://doi.org/10.1007/s40778-014-0008-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-014-0008-7