
Abstract
Renal tubules play an important role in maintaining water, electrolyte, and acid–base balance. Renal tubule dysfunction can cause electrolyte disorders and acid–base imbalance. Clinically, hypokalemic renal tubular disease is the most common tubule disorder. With the development of molecular genetics and gene sequencing technology, hereditary renal tubular diseases have attracted attention, and an increasing number of pathogenic genes related to renal tubular diseases have been discovered and reported. Inherited renal tubular diseases mainly occur due to mutations in genes encoding various specific transporters or ion channels expressed on the tubular epithelial membrane, leading to dysfunctional renal tubular reabsorption, secretion, and excretion. An in-depth understanding of the molecular genetic basis of hereditary renal tubular disease will help to understand the physiological function of renal tubules, the mechanism by which the kidney maintains water, electrolyte, and acid–base balance, and the relationship between the kidney and other systems in the body. Meanwhile, understanding these diseases also improves our understanding of the pathogenesis of hypokalemia, alkalosis and other related diseases and ultimately promotes accurate diagnostics and effective disease treatment. The present review summarizes the most common hereditary renal tubular diseases (Bartter syndrome, Gitelman syndrome, EAST syndrome and Liddle syndrome) characterized by hypokalemia and alkalosis. Further detailed explanations are provided for pathogenic genes and functional proteins, clinical manifestations, intrinsic relationship between genotype and clinical phenotype, diagnostic clues, differential diagnosis, and treatment strategies for these diseases.



Similar content being viewed by others
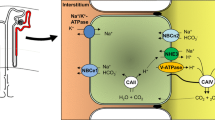
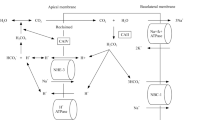

References
Downie ML, Lopez Garcia SC, Kleta R, Bockenhauer D (2021) Inherited tubulopathies of the kidney: insights from genetics. Clin J Am Soc Nephrol 16(4):620–630. https://doi.org/10.2215/CJN.14481119
Zelikovic I (2001) Molecular pathophysiology of tubular transport disorders. Pediatr Nephrol 16(11):919–935. https://doi.org/10.1007/s004670100671
Cunha TDS, Heilberg IP (2018) Bartter syndrome: causes, diagnosis, and treatment. Int J Nephrol Renovasc Dis 11:291–301. https://doi.org/10.2147/IJNRD.S155397
Calò L, Davis PA, Semplicini A (2002) Reduced content of alpha subunit of Gq protein content in monocytes of Bartter and Gitelman syndromes: relationship with vascular hyporeactivity. Kidney Int 61(1):353–354. https://doi.org/10.1046/j.1523-1755.2002.00128.x
Nunez-Gonzalez L, Carrera N, Garcia-Gonzalez MA (2021) Molecular basis, diagnostic challenges and therapeutic approaches of Bartter and Gitelman syndromes: a primer for Clinicians. Int J Mol Sci 22(21):11414. https://doi.org/10.3390/ijms222111414
Konrad M, Nijenhuis T, Ariceta G et al (2021) Diagnosis and management of Bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int 99(2):324–335. https://doi.org/10.1016/j.kint.2020.10.035
Schlingmann KP, Konrad M, Jeck N et al (2004) Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 350(13):1314–1319. https://doi.org/10.1056/NEJMoa032843
Seyberth HW, Koniger SJ, Rascher W, Kuhl PG, Schweer H (1987) Role of prostaglandins in hyperprostaglandin E syndrome and in selected renal tubular disorders. Pediatr Nephrol 1(3):491–497. https://doi.org/10.1007/BF00849259
Kömhoff M, Laghmani K (2017) Pathophysiology of antenatal Bartter’s syndrome. Curr Opin Nephrol Hypertens 26(5):419–425. https://doi.org/10.1097/mnh.0000000000000346
Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP (1996) Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13(2):183–188. https://doi.org/10.1038/ng0696-183
Markadieu N, Delpire E (2014) Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch 466(1):91–105. https://doi.org/10.1007/s00424-013-1370-5
de la Gómez FC, Novoa PJ, Caviedes RN (2019) Bartter syndrome: An infrequent tubulopathy of prenatal onset. Rev Chil Pediatr 90(4):437–442. https://doi.org/10.32641/rchped.v90i4.932
Starremans PG, Kersten FF, Knoers NV, van den Heuvel LP, Bindels RJ (2003) Mutations in the human Na-K-2Cl cotransporter (NKCC2) identified in Bartter syndrome type I consistently result in nonfunctional transporters. J Am Soc Nephrol 14(6):1419–1426. https://doi.org/10.1097/01.asn.0000064948.39199.a0
Kleta R, Bockenhauer D (2006) Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol 104(2):73–80. https://doi.org/10.1159/000094001
Amirlak I, Dawson KP (2000) Bartter syndrome: an overview. QJM 93(4):207–215. https://doi.org/10.1093/qjmed/93.4.207
Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP (1996) Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel. ROMK Nat Genet 14(2):152–156. https://doi.org/10.1038/ng1096-152
Bichet DG, Fujiwara TM (2004) Reabsorption of sodium chloride–lessons from the chloride channels. N Engl J Med 350(13):1281–1283. https://doi.org/10.1056/NEJMp048026
Welling PA, Ho K (2009) A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 297(4):F849-863. https://doi.org/10.1152/ajprenal.00181
Finer G, Shalev H, Birk OS, Galron D, Jeck N, Sinai-Treiman L, Landau D (2003) Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr 142(3):318–323. https://doi.org/10.1067/mpd.2003.100
Simon DB, Bindra RS, Mansfield TA et al (1997) Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet 17(2):171–178. https://doi.org/10.1038/ng1097-171
Seys E, Andrini O, Keck M et al (2017) Clinical and Genetic Spectrum of Bartter Syndrome Type 3. J Am Soc Nephrol 28(8):2540–2552. https://doi.org/10.1681/ASN.2016101057
Han Y, Lin Y, Sun Q, Wang S, Gao Y, Shao L (2017) Mutation spectrum of Chinese patients with Bartter syndrome. Oncotarget 8(60):101614–101622. https://doi.org/10.18632/oncotarget.21355
Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R (2002) X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 415(6869):287–294. https://doi.org/10.1038/415287a
Konrad M, Vollmer M, Lemmink HH et al (2000) Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol 11(8):1449–1459. https://doi.org/10.1681/asn.V1181449
Estévez R, Boettger T, Stein V et al (2001) Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature 414(6863):558–561. https://doi.org/10.1038/35107099
Birkenhäger R, Otto E, Schürmann MJ et al (2001) Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 29(3):310–314. https://doi.org/10.1038/ng752
Nozu K, Inagaki T, Fu XJ et al (2008) Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet 45(3):182–186. https://doi.org/10.1136/jmg.2007.052944
Laghmani K, Beck BB, Yang SS et al (2016) Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med 374(19):1853–1863. https://doi.org/10.1056/NEJMoa1507629
Vargas-Poussou R, Huang C, Hulin P et al (2002) Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol 13(9):2259–2266. https://doi.org/10.1097/01.asn.0000025781.16723.68
Kömhoff M, Laghmani K (2018) MAGED2: a novel form of antenatal Bartter’s syndrome. Curr Opin Nephrol Hypertens 27(4):323–328. https://doi.org/10.1097/mnh.0000000000000422
Carmosino M, Gerbino A, Hendy GN et al (2015) NKCC2 activity is inhibited by the Bartter’s syndrome type 5 gain-of-function CaR-A843E mutant in renal cells. Biol Cell 107(4):98–110. https://doi.org/10.1111/boc.201400069
Besouw MTP, Kleta R, Bockenhauer D (2020) Bartter and Gitelman syndromes: questions of class. Pediatr Nephrol 35(10):1815–1824. https://doi.org/10.1007/s00467-019-04371-y
Simon DB, Nelson-Williams C, Bia MJ, Ellison D et al (1996) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12(1):24–30. https://doi.org/10.1038/ng0196-24
Knoers NV, Levtchenko EN (2008) Gitelman syndrome. Orphanet J Rare Dis 2008(3):22. https://doi.org/10.1186/1750-1172-3-22
Hsu YJ, Yang SS, Chu NF, Sytwu HK, Cheng CJ, Lin SH (2009) Heterozygous mutations of the sodium chloride cotransporter in Chinese children: prevalence and association with blood pressure. Nephrol Dial Transplant 24(4):1170–1175. https://doi.org/10.1093/ndt/gfn619
Kondo A, Nagano C, Ishiko S et al (2021) Examination of the predicted prevalence of Gitelman syndrome by ethnicity based on genome database. Sci Rep 11(1):16099. https://doi.org/10.1038/s41598-021-95521-6
Tago N, Kokubo Y, Inamoto N, Naraba H, Tomoike H, Iwai N (2004) A high prevalence of. Gitelman’s syndrome mutations in Japanese. Hypertens Res 27(5):327–331. https://doi.org/10.1291/hypres.27.327
Lin SH, Shiang JC, Huang CC et al (2005) Phenotype and genotype analysis in Chinese patients with Gitelman’s syndrome. J Clin Endocrinol Metab 90(5):2500–2507. https://doi.org/10.1210/jc.2004-1905
Mastroianni N, De Fusco M, Zollo M et al (1996) Molecular cloning, expression pattern, and chromosomal localization of the human Na-Cl thiazide-sensitive cotransporter (SLC12A3). Genomics 35(3):486–493. https://doi.org/10.1006/geno.1996.0388
Miao M, Zhao CQ, Wang XL, Shan ZY (2016) Clinical and genetic analyses of Chinese patients with Gitelman syndrome. Genet Mol Res. https://doi.org/10.4238/gmr.15027859
Blanchard A, Bockenhauer D, Bolignano D et al (2017) Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 91(1):24–33. https://doi.org/10.1016/j.kint.2016.09.046
Ji W, Foo JN, O’Roak BJ et al (2008) Rare independent mutations in renal salt handling genes. Contribute to blood pressure variation. Nat Genet 40(5):592–599. https://doi.org/10.1038/ng.118
de Baaij JH (2015) The art of magnesium transport. Magnes Res 28(3):85–91. https://doi.org/10.1684/mrh.2015.0388
Nijenhuis T, Vallon V, van der Kemp AW et al (2005) Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115(6):1651–1658. https://doi.org/10.1172/JCI24134
Mabillard H, Sayer JA (2019) The molecular genetics of gordon syndrome. Genes (Basel) 10(12):986. https://doi.org/10.3390/genes10120986
Wilson FH, Disse-Nicodème S, Choate KA et al (2001) Human hypertension caused by mutations in WNK kinases. Science 293(5532):1107–1112. https://doi.org/10.1126/science.1062844
Sethar GH, Almoghawi A, Khan N, Altourah W, Ashour NM (2018) Pseudohypoaldosteronism type II: a young girl presented with hypertension, hyperkalemia and metabolic acidosis. J Coll Physicians Surg Pak 28(3):S21–S22. https://doi.org/10.29271/jcpsp.2018.03.S21
Gordon RD, Geddes RA, Pawsey CG, O’Halloran MW (1970) Hypertension and severe hyperkalaemia associated with suppression of renin and aldosterone and completely reversed by dietary sodium restriction. Australas Ann Med 19(4):287–294. https://doi.org/10.1111/imj.1970.19.4.287
Celmina M, Micule I, Inashkina I et al (2019) EAST/SeSAME syndrome: review of the literature and introduction of four new Latvian patients. Clin Genet 95(1):63–78. https://doi.org/10.1111/cge.13374
Abdelhadi O, Iancu D, Stanescu H, Kleta R, Bockenhauer D (2016) EAST syndrome: Clinical, pathophysiological, and genetic aspects of mutations in KCNJ10. Rare Dis 4(1):e1195043. https://doi.org/10.1080/21675511.2016.1195043
Bockenhauer D, Feather S, Stanescu HC et al (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360(19):1960–1970. https://doi.org/10.1056/NEJMoa0810276
Scholl UI, Choi M, Liu T et al (2009) Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106(14):5842–5847. https://doi.org/10.1073/pnas.0901749106
Botero-Velez M, Curtis JJ, Warnock DG (1994) Brief report: Liddle’s syndrome revisited–a disorder of sodium reabsorption in the distal tubule. N Engl J Med 330(3):178–181. https://doi.org/10.1056/NEJM199401203300305
Liu K, Qin F, Sun X et al (2018) Analysis of the genes involved in Mendelian forms of low-renin hypertension in Chinese early-onset hypertensive patients. J Hypertens 36(3):502–509. https://doi.org/10.1097/hjh.0000000000001556
Yang KQ, **ao Y, Tian T, Gao LG, Zhou XL (2014) Molecular genetics of Liddle’s syndrome. Clin Chim Acta 436:202–206. https://doi.org/10.1016/j.cca.2014.05.015
Muriithi AK, Leung N, Valeri AM et al (2014) Biopsy-proven acute interstitial nephritis, 1993–2011: a case series. Am J Kidney Dis 64(4):558–566. https://doi.org/10.1053/j.ajkd.2014.04.027
Kidder D, Rutherford E, Kipgen D et al (2015) Kidney biopsy findings in primary Sjogren syndrome. Nephrol Dial Transplant 30(8):1363–1369. https://doi.org/10.1093/ndt/gfv042
Berg P, Jeppesen M, Leipziger J (2021) Cystic fibrosis in the kidney: new lessons from impaired renal HCO3-excretion. Curr Opin Nephrol Hypertens 30(4):437–443. https://doi.org/10.1097/MNH.0000000000000725
Lucarelli M, Bruno SM, Pierandrei S et al (2016) The impact on genetic testing of mutational patterns of CFTR gene in different clinical macrocategories of cystic fibrosis. J Mol Diagn 18(4):554–565. https://doi.org/10.1016/j.jmoldx.2016.02.007
Funder JW, Carey RM, Mantero F et al (2016) The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 101(5):1889–1916. https://doi.org/10.1210/jc.2015-4061
Palmer BF, Clegg DJ (2016) Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 40(4):480–490. https://doi.org/10.1152/advan.00121.2016
Palmer BF, Clegg DJ (2019) Physiology and pathophysiology of potassium homeostasis: core curriculum 2019. Am J Kidney Dis 74(5):682–695. https://doi.org/10.1053/j.ajkd.2019.03.427
Palmer BF, Clegg DJ (2019) The use of selected urine chemistries in the diagnosis of kidney disorders. Clin J Am Soc Nephrol 14(2):306–316. https://doi.org/10.2215/cjn.10330818
Elborn JS (2016) Cystic fibrosis. Lancet 388(10059):2519–2531. https://doi.org/10.1016/s0140-6736(16)00576-6
Williams SN, Nussbaum E, Chin TW et al (2014) Diagnosis of cystic fibrosis in the kindred of an infant with CFTR-related metabolic syndrome: importance of follow-up that includes monitoring sweat chloride concentrations over time. Pediatr Pulmonol 49(3):E103-108. https://doi.org/10.1002/ppul.22918
Kose M, Pekcan S, Ozcelik U et al (2008) An epidemic of pseudo-Bartter syndrome in cystic fibrosis patients. Eur J Pediatr 167(1):115–116. https://doi.org/10.1007/s00431-007-0413-3
Suleyman H, Cadirci E, Albayrak A, Halici Z (2008) Nimesulide is a selective COX-2 inhibitory, atypical non-steroidal anti-inflammatory drug. Curr Med Chem 15(3):278–283. https://doi.org/10.2174/092986708783497247
Knoers NV (2006) Gitelman syndrome. Adv Chronic Kidney Dis 13(2):148–154. https://doi.org/10.1053/j.ackd.2006.01.014
Ranade VV, Somberg JC (2001) Bioavailability and pharmacokinetics of magnesium after administration of magnesium salts to humans. Am J Ther 8(5):345–357. https://doi.org/10.1097/00045391-200109000-00008
Yang KQ, Lu CX, Fan P et al (2018) Genetic screening of SCNN1B and SCNN1G genes in early-onset hypertensive patients helps to identify Liddle syndrome. Clin Exp Hypertens 40(2):107–111. https://doi.org/10.1080/10641963.2017.1334799
Acknowledgements
We would like to thank Yuan Liang for the overall polishing of the article.
Funding
This work was supported by a Grant (82174115) from the National Natural Science Foundation of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethical statement
For this type of study (i.e., review article) ethical approval is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guo, W., Ji, P. & **e, Y. Genetic diagnosis and treatment of hereditary renal tubular disease with hypokalemia and alkalosis. J Nephrol 36, 575–591 (2023). https://doi.org/10.1007/s40620-022-01428-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-022-01428-4