
Abstract
Numerous physiological processes happening in the human body, including cerebral development and function, require the participation of biometal ions such as iron, copper, and zinc. Their dyshomeostasis may, however, contribute to the onset of Alzheimer’s disease (AD) and potentially other neurodegenerative diseases. Chelation of biometal ions is therefore a therapeutic strategy against AD. This review provides a survey of natural and synthetic chelating agents that are or could potentially be used to target the metal hypothesis of AD. Since metal dyshomeostasis is not the only pathological aspect of AD, and the nature of this disorder is very complex and multifactiorial, the most efficient therapeutics should target as many neurotoxic factors as possible. Various coumarin derivatives match this description and apart from being able to chelate metal ions, they exhibit the capacity to inhibit cholinesterases (ChEs) and monoamine oxidase B (MAO-B) while also possessing antioxidant, anti-inflammatory, and numerous other beneficial effects. Compounds based on the coumarin scaffold therefore represent a desirable class of anti-AD therapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Chelation of excess biometal ions (Fe, Cu, and Zn) limits their binding to amyloid plaques and the overall aggregation of amyloid beta in the brain, while also reducing oxidative stress. |
Numerous natural and synthetic metal chelators represent potential anti-AD therapeutics. |
Coumarin derivatives exhibit multiple beneficial anti-AD effects including metal chelating activity. |
1 Introduction
AD is a progressive neurodegenerative disease (ND) and the main cause of dementia and death in the elderly. It is characterized by cognitive decline accompanied with consecutive memory loss, depression, and communication problems, all of which are caused by neuronal damage in specific areas of the brain that are responsible for the aforementioned cognitive functions [1]. As the nature of AD is complex and multifactorial, the exact etiology of the disease remains unknown. However, disruption of cholinergic neurotransmission caused by elevated acetylcholinesterase (AChE) activity and the consecutive decline in acetylcholine levels, as well as aggregation of hyperphosphorylated τ-protein, formation of extracellular β-amyloid (Aβ) plaques, oxidative stress, and, finally, dyshomeostasis of biometal ions, are all considered to be the main biopathological aspects of AD. It has been discovered that incorrect folding of Aβ is considerably influenced by the presence of metal ions localized inside as well as on the periphery of amyloid plaques [2]. This has led to an increased interest in the study of metal ion dyshomeostasis as an important factor in AD pathogenesis.
Restoring the correct equilibrium of metal ions in the brain may lead to prevention of Aβ aggregation, decomposition of amyloid plaques (disaggregation of Aβ) and deceleration of the whole process of cognitive decline in patients with AD [3]. Chelation is an effective strategy for restoring the homeostasis of biometal ions with the application of chelators—molecules able to scavenge and bind metal ions through formation of multiple coordinate bonds [4]. Among the diverse metal chelators that have been recently explored to regulate metal homeostasis as a key target in AD therapy [4, 5] are also numerous natural and synthetic coumarin derivatives. These compounds have exhibited a broad spectrum of biological activity, including the ability to inhibit ChEs and MAO-B depending on substituents present, making coumarin a privileged scaffold when designing novel multitarget-directed therapeutics for AD [6].
2 Metal Chelators for the Potential Treatment of AD
The importance of metal dyshomeostasis in the pathogeneais of AD leads to the idea that restoring this homeostasis could prove beneficial in AD treatment [7]. Metal chelators capture metal ions and displace them from their target sites, thus intercepting their participation in redox processes and Aβ aggregation. Chelators either possess the ability to reduce the total extracellular amount of bioavailable metals or act as ionophores, competing with endogenous ligands for metal ions [8].
8-Hydroxyquinoline (8-HQ, 1, Fig. 1), a natural lipophilic molecule, shows strong anti-AD effects via chelation of metal ions [9]. Clioquinol (CQ, 2), a derivative of 8-HQ and a moderate chelator of Fe, Cu, and Zn [10], entered phase II clinical trials as a potential drug for AD [11]. CQ decreased Aβ aggregation and improved cognitive behavior, which was, however, accompanied by severe side effects, and futher clinical trials of CQ were therefore abandoned [12]. PBT2 (3) promotes Aβ degradation and GSK-3 phosphorylation through its chelatory activity [13], while also lowering H2O2 formation [12]. Despite its initial success and promising results in animal models, repeated phase IIb clinical trials of PBT2 ultimately provided no evidence of cognitive improvement in patients with AD as compared with controls [14, 15]. Deferiprone (DFP, 4), an important iron chelator, displays a neuroprotective effect by reducing the level of Aβ and τ phosphorylation [16]. Trientine (TETA, 5), a tetradentate ligand, exhibits β-secretase inhibition, while slowing amyloidosis in mice [17]. TM (6), a conjugate of TETA and melatonin significantly lowered the deposition of Aβ and neuronal degeneration in the brains of APP/PS1 double transgenic mice, while also blocking τ hyperphosphorylation, γ-secretase, oxidative stress, and metal dyshomeostasis [18]. d-Penicillamine (7), a Cu/Zn chelator, can delay AD by reducing oxidative stress in blood serum [19]. Deferoxamine (DFO, 8), preferentially an iron chelator, impaired translation of the amyloid precursor protein (APP) holoprotein and significantly improved certain cognitive abilities in patients with AD [20]. After 3 months of experiments on Tg2576 mice, DP-109 (9) induced an increase in Aβ solubility in the brain, thereby reducing the amount of Aβ aggregates [21]. Additionally, other chelators (e.g., metformin and cyclodipeptides) have effectively been applied to treat NDs, including AD, albeit being originally designed for other purposes [22, 23].

Structure of various metal chelators (1–9)
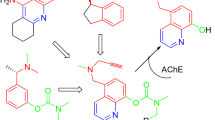
Tenuazonic acid (TA, 10) is a natural compound that exhibits antioxidant and metal chelating properties apart from acting as an AChE inhibitor [24]. Poliseno et al. [25] have synthesized five modern conjugates (11–15, Fig. 2) of TA and donepezil with the aim of creating novel multitarget anti-AD ligands. Compound 11 was shown to be a potent chelator of Fe(III), Cu(II), and Zn(II) (pFe = 16.6, pCu = 11.6, and pZn = 6.0 at pH 7.4). Additionally, all five derivatives were capable of AChE and Aβ aggregation inhibition, while remaining noncytotoxic against SH-SY5Y human neuroblastoma cells. Another recent study by Kilic et al. [26] was focused on the synthesis of novel multifunctional thiourea and benzamide derivatives and investigation of their anti-AD potential (ChEs inhibition, radical scavenging and metal chelation). From this series, the most effective derivatives 16–20 are displayed on Fig. 2. All five compounds were capable of notable Fe(II), Cu(II), and Zn(II) chelation and butyrylcholinesterase (BChE) inhibition. Derivative 20 also displayed AChE-inhibitory activity and considerable antioxidant properties, thus being the most promising anti-AD candidate from the series.

Structure of recently reported chelators (10−20)
2.1 Iron Chelation
Iron can be present in the body in a free form, which is exceptionally toxic, as well as in a bound form, which is beneficial and has a crucial role in maintaining physiological processes in the body. Iron is located in tissues and plasma, where it is bound to transferrin under physiological conditions. However, in some cases, the free form of iron becomes abundant [27]. The most harmful unbound form of iron is the labile plasma iron, which has redox activity, can permeate into organs and induce iron overload [28]. Another form of iron is present in the tissues—tissue ferritin and labile iron pool (LIP). A major amount of iron in the human body is heme-bound in molecules such as hemoglobin and myoglobin. Apart from heme, it is also present in ferritin and hemosiderin, which are responsible for the storage of iron in cells [27]. Iron is mostly bound to proteins and only less than 1% is accessible for physiological (metabolism), toxicological (redox), or pharmacological (chelation) uses [29]. There are no proteins that bind the labile form of iron. In general terms, LIP is characterized as a cellular chelatable pool, which includes various ligand-associated ionic forms of iron. These ligands may include polypeptides, organic anions, and membrane surface components [30]. Considering neurodegenerative changes, the active form of iron plays a crucial role in the mechanisms of oxidation, Aβ plaques formation, etc. LIP exists in both cellular and extracellular forms, while the division into these two categories is based on their propensity to trigger redox processes and the accessibility of the forms to be chelated. LIP generally includes those forms of iron that can participate in redox cycles while also being scavengeable by permeant chelators. Due to the nature of LIP, these forms of iron may have a role in the generation of oxidative stress. Moreover, a significant impact of increasing LIP amounts on reactive oxygen species (ROS) production, lipid peroxidation, and cell death has been demonstrated. The unstable nature of LIP accounts for its high reactivity and subsequent ROS formation [27].
2.1.1 Iron Chelation Therapy
Since iron is involved in the pathogenesis of NDs, iron chelation presents a possible therapeutic strategy. Iron chelation is clinically used in hemochromatosis and iron overdose during blood transfusion [31]. Treatment of these pathological conditions requires high-affinity selective iron chelators that aid in mass excretion of iron from the body [32]. DFO, DFP, and deferasirox (DFX 21, Fig. 3) have been tested as drugs for such conditions [33].

Structure of deferasirox (21), tacrine-deferiprone hybrids (22−29), and natural chelators (30–34)
Several iron chelators that can cross the blood–brain barrier (BBB) have already been explored in the treatment of AD and their neuroprotective mechanisms have been linked to suppression of apoptotic pathways [34], promoting survival pathways [35], restoration of protein degradation [36], and stabilization of mitochondrial function [37]. CQ has a moderate affinity for iron and is capable of Fe chelation [38]. The therapeutic effect of CQ is often attributed to its ionophoric activity, which causes redistribution of copper and zinc to the cell [13, 39, 40]. However, the ability to chelate iron also accounts for its neuroprotective properties. Iron binds to CQ, and many beneficial effects of CQ are iron dependent [32]. Administration of CQ to MPTP-treated mice protects against iron overload in substantia nigra [41] and prevents its age-related degeneration in τ-knockout mice [38], which confers neuroprotection.
DFO, DFP, and DFX are Food and Drug Administration (FDA)-approved iron chelators capable of crossing the BBB [42]. Recently, it has been shown that these agents can lower iron accumulation in patients with AD [43]. DFO binds iron in a 1:1 ratio and is a hexadentate ligand [44]. Although being the most popular iron chelator with more than 30 years of clinical experience [45], DFO application is limited by its short half-life and the need to be administered intravenously or subcutaneously [46]. Some studies have, however, validated the use of intranasal DFO with the aim of reversing iron-induced memory problems by inhibition of Aβ aggregation, neuronal ferroptosis, τ phosphorylation, and aberrant APP cleavage in AD mouse models [47,48,49,50]. DFP, an oral bidentate chelator that binds iron in a 3:1 ratio [51] had beneficial effects against iron accumulation associated with neurodegeneration [52]. A significant decrease in mouse brain iron and insoluble τ polymer levels after DFP administration was observed in a study by Rao et al. [53]. DFP was also conjugated with tacrine by Chand et al. [54] to obtain novel hybrids 22–29 (Fig. 3) that were capable of AChE and Aβ aggregation inhibition, free radical scavenging, and chelation of Cu and Zn in addition to Fe. Finally, DFX is a tridentate oral chelator, binding iron in a 2:1 ratio [55] that was used by Banerjee et al. [56] to block age-related iron accumulation and to reverse altered Aβ metabolism, oxidative stress, and neuroinflammation in rat brains. Another study [57] has indicated that DFX may chelate the iron that aggregates τ or bind τ directly, thus inhibiting its aggregation.
2.1.2 Natural Substances as Iron Chelators
In addition to CQ and other compounds mentioned in previous sections, the interaction of natural substances (Fig. 3) curcumin (30), (−)-epigallocatechin (EGC, 31), (−)-epicatechin-3-gallate (ECG, 32) and (−)-epigallocatechin-3-gallate (EGCG, 33) with metal ions and their chelating abilities were also studied [58].
The hippocampus is known to be one of the most affected areas of the brain in early stages of AD. Neurotoxic heavy metals such as lead and cadmium disrupt cell structure in this area as well [58]. Neurotoxicity in the hippocampal area of rats caused by Pb and Cd was considerably lowered by curcumin [59, 60]. Furthermore, promotion of hippocampal neurogenesis by curcumin was observed in chronically stressed rats [61]. The neuroprotective effect of curcumin is presumably mediated through its ability chelate harmful metal ions, resulting in the formation of solid, inactive complexes. The anti-inflammatory effect of curcumin may also limit nervous tissue swelling [58]. Potential reaction centers of curcumin are the phenolic hydroxyls, the carbonyl groups, and the methylene group in the α-position to the carbonyls. The phenolic and enolic hydroxyl groups are capable of forming complexes with metal ions [62]. These complexes exhibit efficient radical scavenging which, together with the binding of redox-active metals such as Fe and Cu, implies the considerable neuroprotective effect of curcumin. The interaction of curcumin with Fe reaches half of the maximum value at a concentration of 2.5–5 μM and interaction with Cu at a concentration of 3–12 μM [63]. Importance of the curcumin scaffold in the design of new fluorescent probes for AD diagnosis and therapy has also been described [64].
Another potential group of natural iron chelators are polyphenols, which also display antioxidant and anti-inflammatory effects that account for their neuroprotective activity [65]. The ingredients of green tea have been proposed to treat various cardiovascular and inflammatory diseases and some NDs [66, 67]. The major components of green tea are the polyphenolic compounds EGC, ECG, and EGCG [68], with the latter being a relatively potent chelator of both iron and copper [58]. EGC and ECG have been shown to primarily chelate Cu2+ and Zn2+ ions and diminish Cu/Zn-induced or self-assembled Aβ aggregates while also decreasing ROS production [69]. Since the pathogenesis of AD is associated with metal accumulation, tea polyphenols and curcumin may gain significance as potential drugs for AD.
Mandel et al. [70] have actually proposed the use of curcumin and EGCG in the treatment of AD. Both substances are able to cross the BBB and possess multifunctional activities, which include metal chelation, radical scavenging, neuroprotection, and anti-inflammatory activity. The safety and dietary abundance of these compounds further support the interest in their use as anti-AD drugs. The authors hypothesized that these two substances may primarily act as iron chelators, again suggesting an important role of iron in the pathogenesis and progression of AD.
Jahanshahi et al. [71] have studied the effects of another natural flavanone compound naringin (34) on the hippocampus of iron-overloaded mice. Their results showed that naringin has a strong iron chelation capacity and could also decrease the formation of amyloid plaques, which could be beneficial for neuroprotection and AD prevention.
2.2 Copper Chelation
Copper is essential for the proper functioning of organisms, but at increased concentrations it becomes toxic. Thus, its level and bioavailability in the body are very important factors. The majority of copper is bound to proteins or other macromolecules in the body. Nevertheless, the redox activity of copper can lead to highly toxic products. Abnormal and uncontrolled intracellular Cu2+ concentrations contribute to oxidative stress, as the resulting ROS can lead to DNA damage and oxidation of lipids and proteins [72].
On the other hand, superoxide anions, which belong to ROS, are disproportionated in the body by the enzyme Cu/Zn-superoxide dismutase (Cu/Zn-SOD), which uses copper as its catalytic active center. The protection of cells from copper toxicity is mediated by metallothioneins which control the levels of copper ions in the cytoplasm and chaperones that transport them to destined cellular structures [72]. In the blood, copper is bound and transported by albumin and ceruloplasmin, with the latter accounting for 90% of copper transport. Albumin binds Cu2+ ions, which are reduced to Cu+ in the presence of ascorbate. However, molecular oxygen easily reoxidizes Cu+ back to Cu2+ [73]. The “free copper pool” represents a small percentage of copper (5–15%) that is loosely associated with albumin or other small molecules but not covalently bound to ceruloplasmin. The free copper pool must be maintained at low levels, as there is ample evidence that the higher its level, the greater the damage caused by copper toxicity [72].
2.2.1 Copper Chelators and Treatment of AD
Tegoni et al. [72] have described the structure and biological activity of numerous copper chelators. CQ chelates biometal ions through its N and O donor atoms [74] and can cross the BBB. Upon entering the brain tissue, CQ is able to cause partial disaggregation of the Cu-induced Aβ aggregates due to its high affinity for Cu2+ [75]. The chemical mechanism of this activity presumably involves partial resolubilization of Cu-Aβ plaques, inhibition of ROS formation and increased expression of matrix metalloproteinases that degrade Aβ [76, 77]. However, long-term use of CQ has adverse side effects [78] due to the cytotoxicity of elevated levels of copper-CQ complexes (same as in the case of 8-HQ) [74].
Altering the substituents on the same structural motif significantly affects the degree of cytotoxicity of a given compound. The CQ derivative PBT2 displays several advantages over CQ [79, 80], including higher membrane permeability, greater effectiveness in lowering the cerebral level of Aβ plaques and improving cognitive function. PBT2 also possesses fewer side effects than its parent molecule [79, 81]. The probable mechanism of action of CQ and PBT2 involves their permeation across the BBB and reaching the copper-containing Aβ plaques. The chelators compete with aggregated or oligomeric Aβ for Cu2+ binding and form soluble complexes, transporting copper inside the cells and restoring its normal levels by acting as ionophores. The simultaneous activity of matrix metalloproteinases causes the degradation of Aβ peptides [77, 82, 83].
Two novel analogs of CQ—C1 (35) and C2 (36)—have been reported [84,85,86], their structure containing either two 8-aminoquinoline or two 8-hydroxyquinoline skeletons connected via a linker (Fig. 4). These compounds possess the same donor atoms (N and O) as CQ, but exhibit a higher affinity for copper by acting as tetradentate ligands. This accounts for their increased ability to disaggregate Cu-induced Aβ plaques and a remarkable capacity of reducing ROS formation. Moreover, some aromatic chelators, which include bathocuproinedisulfonic acid (37), effectively promote disaggregation of Cu-induced Aβ plaques through an intercalation-like interaction [87].

Synthetic copper chelators (35–37)
2.2.2 Bifunctional Copper Chelators
Bifunctional chelators are ligands that have a second function in addition to possessing a metal binding site. For instance, a reducing group that posseses the ability of inhibiting ROS formation or a moiety supporting the chelator’s active transport across membranes may be incorporated into the main structure. A prodrug preparation strategy has also been presented, in which the chelator molecule contains an activatable or cleavable protecting group that allows it to remain inactive until it reaches its target site, where the chelator is rendered available for metal binding by the action of enzymes or ROS that remove the protecting group. Supplying the chelator with another function is beneficial in terms of specificity, allowing it to operate only when precise conditions are met, and the chelator is located in certain tissues or when specific biomolecules are present [88].
Different aspects affecting bioavailability, toxicity, and efficacy must be considered when designing novel chelators with potential application in clinical AD treatment. The three most necessary factors are: (1) the chelator as well as its corresponding metal complexes must not be toxic, (2) the chelator must display sufficient affinity for the particular metal ion when competing with Aβ but not exceedingly high to not abstract metal cofactors from metalloproteins, and (3) the BBB must be overcome by the drug candidate, which should therefore be appropriately soluble and simultaneously sufficiently lipophilic [88, 89]. These three conditions were met in a number of different methods, which include the incorporation of a metal binding moiety into a molecule capable of interacting with Aβ (incorporation approach) or the linkage of two such components through a spacer (linkage approach) [72].
In the incorporation approach, the aim is to replace some of the original molecule’s carbon atoms by heteroatoms such as N and O in order to obtain a compound that structurally resembles the original molecule. In this way, structures capable of chelating copper and forming five- or six-membered chelate rings are obtainable [72]. Various organic compounds capable of interaction with Aβ aggregates have been studied in the past in order to develop novel pharmaceuticals that target aggregated Aβ or act as probes for in vitro and in vivo imaging of Aβ plaques [90,91,92]. Thioflavine T [90, 91], stilbene, and chalcone derivatives [92, 93], 6-iodo-2-(4-dimethylamino)phenylimidazo[1,2-α]pyridine (IMPY) [94, 95] and diphenylpropyinone [96, 97] were the most commonly used scaffolds in the design of bifunctional chelators 38–60 (Fig. 5).

Bifunctional copper chelators (38−60)
Some of these chelators contain a (N,N)-chelating arrangement that includes two nitrogen atoms, one of which is a part of the pyridine moiety, making them capable of forming 5-membered ring complexes with Cu2+ (38–41, 49–54, 56–57) [72]. Compounds 43 and 44 use pyridyl and carbonyl groups to chelate copper and thus act as (N, O) chelators [97]. In the case of ligands 45–50 and 58–60, six-membered rings are formed during the chelation of Cu2+ [98, 99]. Compound 42 is the only tetradentate ligand in this series [100]. Some of the chelators contain a dimethylamino substituent attached to a benzene or pyridine nucleus (39, 41, 42, 44, and 55–57), which enhances their ability to bind copper–Aβ aggregates. This is attributed to its lipophilic nature and ability to act as a hydrogen bond acceptor [97, 101, 102].
Most of the ligands 38–60 are able to either inhibit the formation of Cu-containing Aβ aggregates or to promote their disaggregation (or both). They do not function as nonspecific chelators but recognize local Cu2+ ions found in Aβ oligomers or aggregates instead [98]. Lim et al. [97, 100,101,102,103,104,105] studied compounds 38–44 and 51–57 in terms of Cu-Aβ aggregation inhibition and disaggregation efficiency. Both Cu-induced and self-induced Aβ1–40 fibrillization still occurs and these chelators are unable to fully suppress aggregation in the presence of Cu2+. However, the ligands cause a significant reorganization and morphological transformations of the aggregates, which results in their shrinkage. A similar behavior was observed for compounds 58–60, which promoted the formation of large amorphous aggregates when coadministered with Cu2+ to Aβ fibrils. Additionally, Aβ aggregation was inhibited and Aβ adducts were partially disaggregated by the action of these three chelators [99].
Furthermore, the aforementioned studies included a series of nuclear magnetic resonance (NMR) experiments to determine whether chelators 38–41, 43, 44, and 51–57 were capable of interaction with Aβ monomers in addition to Aβ aggregates in the presence of a denaturing agent (sodium dodecyl sulfate, SDS) [101,102,103,104,105]. Under such circumstances, Aβ assumes an α-helical conformation, unlike the one present in native conditions [72]. The aggregation of Aβ is reduced as a result of stabilizing its monomeric form by SDS [106]. Chelator administration resulted in altering the chemical shifts of the NH protons in the Aβ chain, with amino acid residues at the N-terminus of the α-helix, near the Cu binding site (His13 and His14), being most affected [101,102,103,104,105]. The interaction of chelators 38–41 and 51–54 with Aβ has also been investigated through molecular docking, which revealed an interaction of the amino, imino and pyridyl groups (H-bond acceptors) of the ligands with the amino acid residues [97, 102,103,104,105]. In the case of ligand 42, NMR evidence revealed its capability of binding to the β-sheet structure of Aβ. This interaction is mediated by specific H-bonds with the backbone of Aβ or by van der Waals forces and π–π interactions between the ligand and lipophilic Aβ residues [100].
Since Cu–Aβ aggregates contribute to ROS production, which might induce oxidative stress and ultimately the neurotoxic aberrations typical for AD [2, 107], the ability to inhibit H2O2 formation was also studied in the case of ligands 38–41 and 55 [102,103,104]. In the presence of these chelators, the amount of H2O2 produced was 30% lower than in their absence. Thus, under the influence of these ligands, not only the structural features of Cu–Aβ aggregates, but also their chemical reactivity is modified. Compounds 42 and 58–60 also displayed significant antioxidant effects [99, 100]. The cytotoxicity of chelators 38–42 and 55 against human neuroblastoma cell lines was also studied, which revealed their cytotoxicity to be very little both in the presence and in the absence of Cu2+ [100, 102,103,104].
In the linkage approach, the Cu-chelating site is designed independently from the main scaffold. The advantage over the incorporation approach resides in the possibility of using a vast variety of Cu-binding moieties and scaffolds targeting Aβ. However, the linkage of two distinct molecular skeletons increases the molar weight and possibly also the hydrophobicity and charge, which results in decreasing the overall bioavailability. The potential number of different bifunctional chelators designed by this method is very large, but only compounds based on thioflavin T and DFP have been prepared and studied so far (Fig. 6) [72].

Bifunctional copper chelators (61−72)
Compounds 61 and 62 contain a bis(pyridylamino) group as a chelating moiety [108, 109], and compound 64 contains a polyaminocarboxylate group [110], while chelators 65–70 are based on DFP [111]. Compound 62 is a tetradentate ligand, and thus complexes Cu2+ better than the tridentate ligand 63 [72]. In analogy with thioflavin T, ligands 61–63 are also capable of interacting with Aβ aggregates and they promote disaggregation of Cu-Aβ adducts. Compound 61 proved to be capable of converting Cu–Aβ1–40 fibrils into a pool of oligomers, while compounds 62 and 63 are able to inhibit the formation of Cu–Aβ1–42 aggregates and cause their disaggregation. The disadvantage of all three compounds lies in their relatively high cytotoxicity [108, 109].
Orvig et al. [111] prepared a series of ligands 65–70 that are based on DFP as a chelating moiety. These ligands contain different substituents, but their activity against Cu-induced Aβ aggregation is almost the same. Interestingly, these ligands selectively inhibited Aβ1-40 aggregation in the presence of Cu2+ but not in the presence of Zn2+. In terms of reactivity related to ROS formation, chelators 62 and 63 are highly potent inhibitors of H2O2 production, with compound 63 being more effective than 62 in this regard [109]. Inhibitory activity on H2O2 production has not been detected in DFP-based chelators, but their ability to scavenge hydroxyl radicals has been demonstrated [111]. In addition, compounds 65–68 display potent antioxidant effects characteristic of DFP [87, 88]. Compound 64 was able to suppress Zn-induced Aβ1–40 aggregation and the same activity can be expected for Cu [110]. It is expected that this molecule could also interact with monomeric Aβ [72]. Ligand 64 is not cytotoxic at millimolar concentrations but is poorly bioavailable in terms of BBB permeability [110].
Vecchio et al. [112] presented a multifunctional Cu2+ chelator 71, which contains a homocarnosine dipeptide (γ-aminobutyryl-l-histidine) connected to the disaccharide trehalose. Trehalose is a potent inhibitor of Aβ aggregation while also being able to reduce its cytotoxicity [113]. Thus, in ligand 71, the homocarnosine unit serves as a binding site for Cu2+, while the function of trehalose is to interact with Aβ. At pH > 6, the compound exhibited exceptional Cu chelation, while also reducing the extensiveness of Aβ aggregation [112]. No evidence has been found on the interaction of compound 71 with Aβ aggregates containing copper, but using trehalose in the linkage approach of novel bifunctional chelators design might prove as a potent strategy [72].
In a recent study by Tan et al. [114], eleven novel 3,3′-diamino-2,2′-bipyridine derivatives were prepared and subsequently tested as potential chelators for AD therapy. The results proved that majority of the derivatives significantly inhibit self-induced aggregation of Aβ1–42. Derivative 72 (Fig. 6) was determined to be most effective in this regard with half-maximal inhibitory concentration (IC50) equal to 9.4 μM and was further subjected to testing its copper chelatory activity. The compound was found to selectively chelate Cu2+ and displayed 66.2% inhibition of Cu2+-induced Aβ1–42 aggregation, while also significantly reversing Aβ-induced memory deficits in mice.
2.2.3 Modern Quinoline-Based Copper Chelators
Although the aforementioned chelators CQ and PBT2 did not ultimately find their application in AD treatment, the quinoline scaffold has remained attractive in terms of designing modern copper chelators with anti-AD activity as observed in a study by Zhao et al. [115] focused on two novel tetradentate chelators TDMQ20 (73) and TDMQ22 (74) (Fig. 7). These derivatives were able to selectively abstract copper from Cu-amyloids without disrupting the activities of tyrosinase or Cu/Zn-SOD and administration of compound 73 to three different murine AD models resulted in a full reversal of their cognitive and behavioral impairment along with reducing oxidative stress in the cortex.

Modern quinoline-based copper chelators (73−80)
Pavlidis et al. [116] have synthesized another series of quinoline-based copper chelators 75–78. Compound 76 exhibited considerable neuroprotection against Aβ- and H2O2-induced toxicities in SH-SY5Y cells, while derivative 77 was more effective against H2O2 after complexation with copper rather than as a sole molecule. According to molecular docking simulations, the authors have additionaly proposed a strong interaction between the two ligands and AChE, indicating that they might also act as inhibitors of the enzyme. Moreover, a recent study [117] describes the synthesis of two novel sulfur-bridged quinoline derivatives 1TPIQ (79) and 2TPQ (80) that were tested synergistically with ascorbic acid (AA) to provide copper chelation while scavenging ROS at the same time. The best synergistic mechanism was observed in a ligand:Cu:AA ratio of 2:1:7.
2.3 Zinc Chelation
Divalent zinc ions are a typical structural or catalytic component of numerous proteins [118]. Apart from Zn2+ ions present in metalloproteins, they are also found in the pool of mobile zinc (mZn), which requires strict regulation in terms of concentration and biochemical activity in order to properly maintain physiological processes and prevent pathological repercussions [119]. The role of mZn in the CNS is particularly important, as the concentration of chelatable zinc is high in specific regions of the brain [120]. The amygdala, hippocampus, and cerebral cortex represent the primary areas of the forebrain where mZn is abundantly located in axons [121]. High levels of mZn can lead to inhibition of protein-tyrosine phosphatases [122], allosteric blockage of NMDA receptors [123], TrkB kinase transactivation [124, 125], and modulation of AMPA receptors function [121, 126].
2.3.1 Zinc Chelation Therapy
Since Aβ aggregation and amyloid plaque deposition in AD is ameliorated by zinc ions, research into the design of new therapeutics has also been focused on the ability of Zn complexation. Indeed, the chelators CQ and DP-109 were found to significantly reduce amyloid plaque formation in the brains of APP/PS1 double transgenic mice [127] and aged female hAbetaPP-transgenic Tg2576 mice [21], respectively. Affecting the toxic oligomerization of Aβ in AD mediated by Cu2+ and Zn2+, PBT2 significantly lowered Aβ amounts in cerebrospinal fluid and improved cognitive abilities in patients with AD [128]. Another potential therapeutic structure is the dipeptide carnosine with Cu/Zn-chelating properties. Carnosine as a dietary supplement has been found to reduce hippocampal intraneuronal Aβ accumulation and prevent mitochondrial dysfunction in triple-transgenic AD mice but has no effect on τ pathology development [129]. Furthermore, bis(thiosemicarbazonato) complexes have been demonstrated to reduce extracellular Aβ levels through selective intracellular zinc delivery [130]. Presenilin, which mediates the proteolytic cleavage of APP, is also eminent for copper/zinc turnover in cells and could indirectly affect Aβ aggregation by reducing Zn2+ levels [131].
The design and optimization criteria for mZn chelators vary, but all must meet certain common requirements regarding their structure, charge, and binding kinetics. Most importantly, they need to exhibit specificity for zinc over other biometal cations [118]. Such specificity is achieved through application of the theory of soft and hard acids and bases [132]. Soft Lewis bases will preferentially bind to soft metal ions (Zn2+) over hard alkali or alkaline earth metal ions (Ca2+). Chelators intended for intracellular administration should be neutral and sufficiently hydrophobic in nature to ensure their passive diffusion across the plasma membrane. In contrast, extracellular chelators generally carry an overall negative charge to minimize their translocation across the cell membrane [118]. Protonation of the donor atoms may limit the rate of chelation, as these protons are required to dissociate first [133]. The chelators must also possess sufficient affinity for mZn to avoid disrupting the homeostasis of Zn2+ ions or the zinc proteome [118].
2.3.2 Intracellular and Extracellular Chelators of Zinc
The most common intracellular zinc chelator is N,N,N′,N′-tetrakis(2-pyridylmethyl)-ethylenediamine (TPEN, 81, Fig. 8). TPEN easily crosses the cell membrane and, using six donor atoms, forms a stable complex with Zn (Kd Zn = 0.7 fM) [118]. The fairly soft nitrogen donor atoms of the pyridine nuclei and amino groups contribute to the selectivity of TPEN for zinc over other, harder divalent cations (e.g., Ca2+). However, TPEN also exhibits high affinity toward other chelatable transition metals (i.e., copper) [118]. Moreover, the toxicity of TPEN has been established, which lies in its ability to induce axon and dendrite degeneration [134], to capture zinc cofactors from metalloproteins [135] and to promote apoptosis [136, 137]. Therefore, research has been focused on the development of advanced zinc chelators as drugs for NDs [138] as well as prochelators capable of crossing the BBB [139] and multifunctional chelators that target specific proteins [103, 140].

Intracellular and extracellular zinc chelators (81−89)
On the other hand, extracellular zinc chelators have been investigated to a much greater extent. The most common zinc extracellular chelators include ethylenediaminetetraacetic acid (EDTA), ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA, 82), 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 83) and ethylenediamine-N, N′-diacetate-N, N′-dipropionate (EDPA, 84) [141, 142]. Although these chelators tightly coordinate Zn2+ through their carboxyl groups, whose anionic nature prevents intracellular transfer, their disadvantage lies in their high affinity for Ca2+ and Mg2+ [118]. To prevent disruption of the extracellular calcium levels, another zinc chelator has been developed—the calcium salt of EDTA, CaEDTA (85). However, CaEDTA displays a significantly lower affinity for Zn (Kd Zn = 2 nM) along with slower zinc binding rate (k = 0.0024 s–1) than EDTA since Ca2+ needs to dissociate prior to zinc binding. The kinetics of CaEDTA chelation are, therefore, relatively slow [141, 142]. As an alternative, the use of tricine (86) as a metal-chelating buffer has been proposed [142]. However, tricine has a low affinity for zinc (Kd Zn = 10 μM) [118].
Nagano et al. [143] used TPEN as a model structure for the synthesis of sodium salts of (4-{[2-bis-pyridin-2-ylmethylamino)ethylamino]methyl}phenyl)methanesulfonic acid (DPESA, 87) and [4-({[2-bis-pyridin-2-ylmethylamino)ethyl]pyridin-2-ylmethylamino}-methyl)phenyl]methanesulfonic acid (TPESA, 88). Both compounds selectively chelate zinc through their nitrogen atoms in a similar fashion to TPEN but are negatively charged owing to the presence of sulfonate groups, making them unable to cross cell membranes. DPESA and TPESA coordinate zinc with picomolar affinities and the rate of Zn2+ chelation is slower for DPESA than TPESA [143]. Still, both ligands 87 and 88 are superior to CaEDTA in terms of zinc chelation [118]. Another synthesis of an extracellular zinc chelator consisting of a dipicolylamine moiety as the major Zn-binding component and aniline-2-sulfonic acid as the bearer of a negative charge was reported [144]. The resulting chelator ZX1 (89) easily binds Zn with a Kd Zn = 1 nM and kZn = 0.027 s–1, which ensures its superiority over CaEDTA [118].
3 Coumarin Derivatives as Metal Chelators
The broad pharmacological activity spectrum of coumarin derivatives is dependent upon their structure and on the substituents present. Among the most studied pharmacological aspects of these compounds, Stefanachi et al. [6] have emphasized their antibacterial, antitubercular, antifungal, antiviral, antimutagenic, antioxidant, anti-inflammatory, antithrombotic, anticancer, anticoagulant, vasodilatory, and cytotoxic effects. They are also able to scavenge ROS and inhibit ChEs. Many of these activities result from the remarkable chemical structure of the oxaheterocyclic system, which enables the molecule to easily bind to numerous target proteins. The coumarin skeleton is characterized by planarity, aromaticity, and lipophilicity, allowing its interaction with lipophilic biomolecular binding sites through strong hydrophobic, but mainly π–π stacking or cation–π interactions with aromatic or positively charged amino acids, respectively. In addition, the lactone group supplies coumarin with the ability to form hydrogen bonds and dipole–dipole interactions [6].
Regardless of the mechanism of action, the binding strength of coumarins to target structures is augmented by additional interactions involving the substituents present on the coumarin scaffold, ultimately establishing their biological activity profile [6]. Concerning the chelatory properties of coumarin itself, Aalikhani et al. [145] have recently shown that coumarin was able to chelate Fe2+ ions in vitro, while also being able to stimulate the activities of superoxide dismutase and catalase in vivo.
3.1 Hydroxycoumarins and Their Derivatives
The simplest natural coumarins are substituted with one hydroxyl group, as observed in the case of 7-hydroxycoumarin (umbelliferone, 90) [146]. DPPH radical scavenging by umbelliferone is concentration dependent [147]. At a concentration of 50 μg ml−1, umbelliferone displays 59.6% inhibition [148], while AA (standard) exhibits 96% inhibition of DPPH [149]. Umbelliferone also scavenges hydroxyl radicals at all concentrations [150], and its inhibition efficiency of membrane reactive hydroxyl radicals is 63.6% [148]. Furthermore, umbelliferone inhibits superoxide anion formation in the oxidation process of NADH by phenazine methosulfate with IC50 of 150 μM compared with AA (IC50 of 86 μM) [146]. In the case of the ABTS+ radical cation, umbelliferone scavenged 68% of the species (AA 80%). The half-maximal effective concentration (EC50) value of umbelliferone (16.7) did not surpass the EC50 value of AA (14.9) [148]. The ability of umbeliferone to chelate Fe2+ ions has also been demonstrated. Umbeliferone reduced Fe3+ to Fe2+ and bound 58.8 % of Fe2+ ions at a concentration of 200 μg ml−1 [150].
Natural coumarin derivatives able to chelate metals and inhibit ROS include other simple hydroxycoumarins and methoxycoumarins (Fig. 9), such as 6,7-dihydroxycoumarin (esculetin, 91), 7,8-dihydroxycoumarin (daphnetin, 92), 7-hydroxy-6-methoxycoumarin (scopoletin, 93), 6,7-dimethoxycoumarin (scoparone, 94), and the glycoside 6-β-d-glucopyranosyloxy-7-hydroxycoumarin (esculin, 95) [147].

Structure of natural coumarin derivatives (90−95)
Mladěnka et al. [147] have studied the copper-chelating activity of compounds 90–95, as well as other coumarin derivatives 96–104 (Fig. 10). All derivatives possessing the ortho-dihydroxy moiety showed the ability to chelate Cu2+. Only minor or no variations in the chelating activities of 6,7-dihydroxycoumarins and 7,8-dihydroxycoumarins were observed. Of the remaining compounds, only derivative 101 was able to chelate Cu2+ cations, although to a much lower extent than o-dihydroxycoumarins. Coumarin derivatives containg different substituents or a 1,3-arrangement of hydroxy groups were almost incapable of chelating copper.

Structure of coumarin derivatives (96−104)
The chelating activity and complex stoichiometry of o-dihydroxycoumarins 91, 92 and 96 were studied in more detail under noncompetitive conditions. The results suggested that the preferred stoichiometric ratio was 2:1, since approximately 50% of copper ions were chelated at a 1:1 ratio of coumarin to Cu2+. All three derivatives were capable of chelating Cu2+ cations in the pH range 5.5–7.5, but at pH 4.5, chelation was no longer observed for either of the compounds. Cu+ ions were not complexed at any pH value. No differences were observed in the chelation activity of derivatives 92 and 96; both compounds formed complexes with Cu2+ ions at a ratio of 2:1. However, derivative 91 coordinated Cu2+ ions at pH 5.5 and 6.8 with a stoichiometry of 1:1, which gradually changed to 2:1 at pH 7.5 [147].
With respect to copper and iron ions, the same functional groups participate in their chelation [147]. In addition to potent o-dihydroxycoumarins, diacetoxycoumarins 101 and 102 were capable of chelating both transition metals, although to a much lower extent due to the electron-withdrawing effect of the carbonyl groups [151]. In the case of iron chelation, the differences between 6,7-dihydroxy and 7,8-dihydroxy groups were more notable as opposed to copper, with the 7,8-dihydroxycoumarins 92 and 96 being the most potent iron chelators with their activity being comparable to DFP [151].
A modern theranostic agent 105 (Fig. 11) based on 4-hydroxycoumarin has recently been presented [152]. Apart from acting as a fluorescence probe, the compound exhibited numerous anti-AD effects, including chelation of various metal ions (mainly Cu2+), ROS elimination (IC50 of 3.10 μM against OH·) and inhibition of Cu2+-induced and self-induced Aβ aggregation, which was more effective when 105 was already coordinated with Cu(II). The ligand also showed low cytotoxicity and was predicted to cross the BBB, making it a very attractive molecule for both the diagnosis and treatment of AD. Another study by the same group [153] describes the synthesis and biological activity of three coumarin derivatives 106–108 that possess a similar structure to 105 and an even better anti-AD activity profile. The compounds were also predicted to cross the BBB, could chelate Cu2+ in a 2:1 ratio, showed stronger OH· scavenging than 105 (IC50 of 1.33–1.54 μM) and even possessed selective inhibitory activity against AChE (IC50 of 11.15–61.47 μM) as opposed to BChE (IC50 > 100 μM). All three derivatives have further displayed certain inhibitory activity against Aβ1–42 aggregation, with 106 and 107 being the most potent ones against self-induced and Cu2+-induced aggregation, respectively. Both studies have demonstrated that such 4-hydroxycoumarins could be a leading class of novel multitarget-directed ligands for the potential treatment of AD. Du et al. [154] have also attempted to elucidate the anti-AD potential of wedelolactone (109) by computational studies. Their results indicated that wedelolactone has exceptionally good affinity to both AChE and BChE, could scavenge hydroperoxyl radicals and act as a potent Cu(II) chelator. However, this is yet to be experimentally confirmed.

Structure of coumarin derivatives (105−109)
3.2 Hydroxypyridinone-Coumarin Hybrids
The design and synthesis of new hydroxypyridinone-coumarin conjugates is based on the idea of linking two pharmacophores, each of which possesses a distinct therapeutic effect against AD, to form multifunctional anti-AD agents. In this case, the aim is to combine the hydroxypyridinone structure which provides good water solubility with coumarin derivatives that are highly lipophilic and cross the BBB well [155]. In terms of biological activity, hydroxypyridinone derivatives have been used as effective, nontoxic iron chelators [156], while coumarins, in addition to their many other beneficial effects [6], display the ability to inhibit MAO-B [157]. MAO-B catalyzes the oxidative deamination of β-phenylethylamines and benzylamines, using FAD as a cofactor. However, the catalytic activity of MAO-B elevates the concentration of neurotoxic species, including aldehydes and H2O2, which contribute to ROS formation and damage neuronal tissue [158, 159]. MAO-B inhibition is therefore one of many desirable effects of anti-AD drugs. MAO-B inhibitors also help to improve memory and learning ability in animals through suppression of oxidative stress [160].
Mi et al. [161] have synthesized a series of hydroxypyridinone-coumarin hybrids 110–120 (Fig. 12). Subsequently, their chelating activity against Fe3+ ions as well as their ability to inhibit human MAO-B were investigated. Compounds 110 and 111 proved to be the most effective chelators with pFe3+ values of 18.9 and 19.8, respectively, thus outperforming DFP used as a standard (pFe3+ = 17.3). However, other derivatives 112–120 also showed good chelating activity with pFe3+ values around 17.

Structure of hydroxypyridinone–coumarin hybrids (110−120)
The inhibitory activity of compounds 110–120 against human MAO-B was tested at two concentrations of the derivatives, 1 μM and 10 μM. Compounds that exhibited significant inhibitory activity were subsequently subjected to the IC50 assay. Introduction of the hydroxypyridinone structure into the parent structure of coumarin did not impair its capacity to inhibit MAO-B. Majority of the derivatives showed a weak inhibitory effect when tested in 1 μM concentrations. However, at 10 μM, their inhibitory potency surpassed 90%. As a result, the IC50 values of all derivatives are less than 10 μM. At 1 μM concentrations, the inhibitory potencies of 110 and 116 were greater than 20%, while compounds 115, 118, and 120 exceeded 30%. Derivatives 111 and 119 were shown to be the most potent MAO-B inhibitors, with 54% and 62% inhibitory effects, respectively. The IC50 values of these two compounds were discovered to be 0.68 μM and 0.86 μM, respectively [161].
In terms of structure-activity relationship, the introduction of a hydroxymethyl substituent at position 2 of the hydroxypyridinone scaffold resulted in a significant improvement in iron chelating activity. Neither this activity, nor the ability to inhibit MAO-B seemed to be affected by the length of the carbon linker connecting the triazole and hydroxypyridinone rings. However, the MAO-B inhibitory activity of 3- and 7-substituted coumarins was significantly higher than that of 4-substituted derivatives [161].
**e et al. [155] have synthesized another series of hydroxypyridinone-coumarin derivatives 121–144 (Fig. 13) and studied their chelating activity as well as MAO-B inhibitory activity. Majority of the derivatives exhibited potent iron-chelating ability (pFe3+ ≈ 18). Derivatives 135 and 139 with pFe3+ values of 18.75 and 18.65, respectively, outperformed the DFP standard (pFe3+ = 17.3) and were the most effective iron chelators from the series. However, all other compounds from the series 121–144 retained the excellent iron-chelating activity of the hydroxypyridinone pharmacophore as well.

Structure of hydroxypyridinone–coumarin hybrids (121−144)
The MAO-B inhibitory activity of compounds 121–144 was measured at a concentration of 10 μM with pargyline being used as a standard MAO-B inhibitor. Despite the incorporation of the hydroxypyridinone scaffold onto the coumarin skeleton, the obtained compounds maintained a significant MAO-B inhibitory activity. Derivatives whose inhibitory activity exceeded 75% were subjected to the IC50 test. Compounds containing a 7-substituted coumarin scaffold proved to be more potent MAO-B inhibitors and their IC50 values ranged from submicromolar to lower nanomolar concentrations. Derivative 136 exhibited the highest MAO-B inhibitory activity (IC50 of 14.7 nM), surpassing even pargyline (IC50 of 85.8 nM) in this regard. Compounds 121–123 were less potent than their respective 7-methoxy (124–126) and 7-propargyloxy (127–129) analogs. Introduction of a benzyloxy group at position 7 led to a further enhancement of the inhibitory activity. After the incorporation of electron-withdrawing substituents (Cl and F) at meta- or para-positions of the benzyloxy phenyl ring, derivatives 133–144 were obtained, which exhibited an even better MAO-B inhibitory activity [155].
Compound 136 was further investigated due to its highest MAO-B inhibitory activity and its cytoprotective effect against oxidative stress as well as its significant ability to improve cognitive abilities in mice have been demonstrated [155]. This derivative also did not exhibit any cytotoxicity and is therefore a promising multifunctional anti-AD drug.
4 Conclusions and Outlook
The treatment of AD has been the subject of interest of numerous scientific studies. New potential anti-AD drugs are constantly being developed and tested for their activity. Owing to the multifactorial nature of the disease, the design of compounds that would target as many stages of the AD neurotoxic cascade as possible, is, however, a great challenge and an enourmously difficult process. It has now been well-established that there is a connection between biometal dyshomeostasis and AD. Therefore, metal chelation may be regarded not only as a therapeutic strategy but also as a way of reducing the risk of AD onset and progression by including natural compounds with anti-AD activity, many of which have been described in this review, in diet.
However, even though extensive research has been conducted in this area over the past decades, there is still no effective chelator therapeutically used in AD treatment. Lack of relevant data and firm conclusions seems to be the limiting factor when concerning the advantages of metal chelation therapy in AD. There is still a need for deeper investigation and extensive clinical studies to provide a more comprehensive view of intracellular metal trafficking and the action of metal chelators in vivo to ultimately establish effective chelators for AD treatment. Moreover, there is a number of distinctive challenges concerning the application of metal chelators for the treatment of NDs. A clinically acceptable chelator must be well absorbed into the bloodstream and capable of crossing the BBB, while binding only to its specific metal ion targets located in distinct areas of the brain in preference to other divalent cations in the rest of the body (e.g., Ca2+ or Mg2+). Nonremoval of specific metals from intracellular deposits, redistibution of toxic metals, and loss of essential metals are all limitations responsible for the mild or serious side effects that a majority of current clinically used chelators exhibit. Toxic chelates may be formed as a consequence of metal binding or a removal of metal ions from essential proteins and cofactors may occur. These numerous challenges and limitations seem to be the prime reason why metal chelators have yet to reach clinical acceptance as anti-AD drugs.
The present review provides a summary of chelating agents that have been isolated and/or synthesized so far with the aim of rebalancing metal dyshomeostasis in the body. Since the coumarin molecule is a desirable scaffold when concerning the design of new potential anti-AD drugs, particularly owing to its peculiar structure and broad spectrum of biological activity, the latter sections of this review are specifically dedicated to coumarin-based metal chelators. Such molecules may provide a novel approach of targeting multiple aspects of the neurotoxic cascade of AD and are a promising class of potential anti-AD therapeutics for further research.
References
Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 2018. https://doi.org/10.1016/j.jalz.2018.02.001.
Savelieff MG, Lee S, Liu Y, Lim MH. Untangling amyloid-beta, Tau, and metals in Alzheimer’s disease. Acs Chem Biol. 2013. https://doi.org/10.1021/cb400080f.
Kawada H, Blessing K, Kiyota T, Woolman T, Winchester L, Kador PF. Effects of multifunctional antioxidants on mitochondrial dysfunction and amyloid-β metal dyshomeostasis. J Alzheimers Dis. 2015. https://doi.org/10.3233/JAD-132471.
Santos MA, Chand K, Chaves S. Recent progress in multifunctional metal chelators as potential drugs for Alzheimer’s disease. Coord Chem Rev. 2016. https://doi.org/10.1016/j.ccr.2016.04.013.
Chaves S, Várnagy K, Santos MA. Recent multi-target approaches on the development of anti-Alzheimer’s agents integrating metal chelation activity. Curr Med Chem. 2021. https://doi.org/10.2174/0929867328666210218183032.
Stefanachi A, Leonetti F, Pisani L, Catto M, Carotti A. Coumarin: a natural, privileged and versatile scaffold for bioactive compounds. Molecules. 2018. https://doi.org/10.3390/molecules23020250.
White AR, Kanninen K, Crouch P. Editorial: Metals and neurodegeneration: restoring the balance. Front Aging Neurosci. 2015. https://doi.org/10.3389/fnagi.2015.00127.
Duce JA, Bush AI. Biological metals and Alzheimer’s disease: implications for therapeutics and diagnostics. Prog Neurobiol. 2010. https://doi.org/10.1016/j.pneurobio.2010.04.003.
Tiffany-Castiglioni E, Hong S, Qian Y. Copper handling by astrocytes: insights into neurodegenerative diseases. Int J Dev Neurosci. 2011. https://doi.org/10.1016/j.ijdevneu.2011.09.004.
Leko MB, Horvat LL, Popovački EŠ, Zubčić K, Hof PR, Šimić G. Metals in Alzheimer’s disease. Biomedicines. 2023. https://doi.org/10.3390/biomedicines11041161.
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, **linas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003. https://doi.org/10.1001/archneur.60.12.1685.
Ejaz HW, Wang W, Lang M. Copper toxicity links to pathogenesis of Alzheimer’s disease and therapeutics approaches. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21207660.
Crouch PJ, Savva MS, Hung LW, Donnelly PS, Mot AI, Parker SJ, Greenough MA, Volitakis I, Adlard PA, Cherny RA. The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J Neurochem. 2011. https://doi.org/10.1111/j.1471-4159.2011.07402.x.
Summers KL, Roseman GP, Sopasis GJ, Millhauser GL, Harris HH, Pickering IJ, George GN. Copper(II) Binding to PBT2 differs from that of other 8-hydroxyquinoline chelators: implications for the treatment of neurodegenerative protein misfolding diseases. Inorg Chem. 2020. https://doi.org/10.1021/acs.inorgchem.0c02754.
Drew SC. Chelator PBT2 forms a ternary Cu2+ complex with β-amyloid that has high stability but low specificity. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms24119267.
Prasanthi JRP, Schrag M, Dasari B, Mararha G, Dickson A, Kirsch WM, Ghribi O. Deferiprone reduces amyloid-β and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J Alzheimers Dis. 2012. https://doi.org/10.3233/JAD-2012-111346.
Wang CY, **e JW, Xu Y, Wang T, Cai JH, Wang X, Zhao BL, An L, Wang ZY. Trientine reduces BACE-1 activity and mitigates amyloidosis via the AGE/RAGE/NF-κB pathway in a transgenic mouse model of Alzheimer’s disease. Antioxid Redox Signal. 2013. https://doi.org/10.1089/ars.2012.5158.
Li LB, Fan YG, Wu WX, Bai CY, Jia MY, Hu JP, Gao HL, Wang T, Zhong ML, Huang XS, Guo C. Novel melatonin-trientine conjugate as potential therapeutic agents for Alzheimer’s disease. Bioorg Chem. 2022. https://doi.org/10.1016/j.bioorg.2022.106100.
Squitti R, Rossini PM, Cassetta E, Moffa F, Pasqualetti P, Cortesi M, Colloca A, Rossi L, Finazzi-Agro’ A. d-Penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur J Clin Investig. 2002. https://doi.org/10.1046/j.1365-2362.2002.00933.x.
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem. 2002. https://doi.org/10.1074/jbc.M207435200.
Lee JY, Friedman JE, Angel I, Kozak A, Koh JY. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiol Aging. 2004. https://doi.org/10.1016/j.neurobiolaging.2004.01.005.
Lanza V, Milardi D, Di Natale G, Pappalardo G. Repurposing copper(II)-chelating drugs for the treatment of neurodegenerative diseases. Curr Med Chem. 2018. https://doi.org/10.2174/0929867324666170518094404.
Yang GJ, Liu H, Ma DL, Leung CH. Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. J Biol Inorg Chem. 2019. https://doi.org/10.1007/s00775-019-01712-y.
Piemontese L, Vitucci G, Catto M, Laghezza A, Perna FM, Rullo M, Loiodice F, Capriati V, Solfrizzo M. Natural scaffolds with multi-target activity for the potential treatment of Alzheimer’s disease. Molecules. 2018. https://doi.org/10.3390/molecules23092182.
Poliseno V, Chaves S, Brunetti L, Loiodice F, Carrieri A, Laghezza A, Tortorella P, Magalhães JD, Cardoso SM, Santos MA, Piemontese L. Derivatives of tenuazonic acid as potential new multi-target anti-Alzheimer’s disease agents. Biomolecules. 2021. https://doi.org/10.3390/biom11010111.
Kilic B, Bardakkaya M, Sagkan RI, Aksakal F, Shakila S, Dogruer DS. New thiourea and benzamide derivatives of 2-aminothiazole as multi-target agents against Alzheimer’s disease: design, synthesis, and biological evaluation. Bioorg Chem. 2023. https://doi.org/10.1016/j.bioorg.2022.106322.
Wojtunik-Kulesza K, Oniszczuk A, Waksmundzka-Hajnos M. An attempt to elucidate the role of iron and zinc ions in development of Alzheimer’s and Parkinson’s diseases. Biomed Pharmacother. 2019. https://doi.org/10.1016/j.biopha.2018.12.140.
Cabantchik ZI, Breuer W, Zanninelli G, Cianciulli P. LPI-labile plasma iron in iron overload. Best Pract Res Clin Haematol. 2005. https://doi.org/10.1016/j.beha.2004.10.003.
Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Body iron metabolism and pathophysiology of iron overload. Int J Hematol. 2008. https://doi.org/10.1007/s12185-008-0120-5.
Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radic Biol Med. 2002. https://doi.org/10.1016/s0891-5849(02)01006-7.
Nick H. Iron chelation, quo vadis? Curr Opin Chem Biol. 2007. https://doi.org/10.1016/j.cbpa.2007.04.025.
Hare D, Ayton S, Bush A, Lei P. A delicate balance: iron metabolism and diseases of the brain. Front Aging Neurosci. 2013. https://doi.org/10.3389/fnagi.2013.00034.
Piga A, Galanello R, Forni GL, Cappellini MD, Origa R, Zappu A. Randomized phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematologica. 2006;91:873–80.
Gal S, Zheng H, Fridkin M, Youdim MBH. Restoration of nigrostriatal dopamine neurons in post-MPTP treatment by the novel multifunctional brain-permeable iron chelator-monoamine oxidase inhibitor drug, M30. Neurotox Res. 2010. https://doi.org/10.1007/s12640-009-9070-9.
Avramovich-Tirosh Y, Bar-Am O, Amit T, Youdim MBH, Weinreb O. Up-regulation of hypoxia-inducible factor (HIF)-1α and HIF-target genes in cortical neurons by the novel multifunctional iron chelator anti-Alzheimer drug, M30. Curr Alzheimer Res. 2010. https://doi.org/10.2174/156720510791162403.
Zhu W, **e W, Pan T, Xu P, Fridkin M, Zheng H, Jankovic J, Youdim MBH, Le W. Prevention and restoration of lactacystin-induced nigrostriatal dopamine neuron degeneration by novel brain-permeable iron chelators. FASEB J. 2007. https://doi.org/10.1096/fj.07-8386com.
Youdim MBH, Fridkin M, Zheng H. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech Ageing Dev. 2005. https://doi.org/10.1016/j.mad.2004.08.023.
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BXW, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012. https://doi.org/10.1038/nm.2613.
Li C, Wang J, Zhou B. The metal chelating and chaperoning effects of clioquinol: insights from yeast studies. J Alzheimers Dis. 2010. https://doi.org/10.3233/jad-2010-100024.
Park MH, Lee SJ, Byun HR, Kim Y, Oh YJ, Koh JY, Hwang JJ. Clioquinol induces autophagy in cultured astrocytes and neurons by acting as a zinc ionophore. Neurobiol Dis. 2011. https://doi.org/10.1016/j.nbd.2011.01.009.
Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitakis I, Ellerby L, Cherny RA, Bush AI, Andersen JK. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron. 2003. https://doi.org/10.1016/s0896-6273(03)00126-0.
Entezari S, Haghi SM, Norouzkhani N, Sahebnazar B, Vosoughian F, Akbarzadeh D, Islampanah M, Naghsh N, Abbasalizadeh M, Deravi N. Iron chelators in treatment of iron overload. J Toxicol. 2022. https://doi.org/10.1155/2022/4911205.
Wang J, Fu J, Zhao Y, Liu Q, Yan X, Su J. Iron and targeted iron therapy in Alzheimer’s disease. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms242216353.
Farr AC, **ong MP. Challenges and opportunities of deferoxamine delivery for treatment of Alzheimer’s disease, Parkinson’s disease, and intracerebral hemorrhage. Mol Pharm. 2021. https://doi.org/10.1021/acs.molpharmaceut.0c00474.
Parker JB, Griffin MF, Downer MA, Akras D, Berry CE, Cotterell AC, Gurtner GC, Longaker MT, Wan DC. Chelating the valley of death: deferoxamine’s path from bench to wound clinic. Front Med. 2023. https://doi.org/10.3389/fmed.2023.1015711.
Schreiner OD, Schreiner TG. Iron chelators as a therapeutic option for Alzheimer’s disease—a mini-review. Front Aging. 2023. https://doi.org/10.3389/fragi.2023.1234958.
Hanson LR, Fine JM, Renner DB, Svitak AL, Burns RB, Nguyen TM, Tuttle NJ, Marti DL, Panter SS, Frey WH. Intranasal delivery of deferoxamine reduces spatial memory loss in APP/PS1 mice. Drug Deliv Transl Res. 2012. https://doi.org/10.1007/s13346-011-0050-2.
Zhu D, Liang R, Liu Y, Li Z, Cheng L, Ren J, Guo Y, Wang M, Chai H, Niu Q, Yang S, Bai J, Yu H, Zhang H, Qin X. Deferoxamine ameliorated Al(mal)3-induced neuronal ferroptosis in adult rats by chelating brain iron to attenuate oxidative damage. Toxicol Mech Methods. 2022. https://doi.org/10.1080/15376516.2022.2053254.
Guo C, Zhang YX, Wang T, Zhong ML, Yang ZH, Hao LJ, Chai R, Zhang S. Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1α pathway on the brain of APP/PS1 transgenic mice. Front Aging Neurosci. 2015. https://doi.org/10.3389/fnagi.2015.00104.
Guo C, Wang P, Zhong ML, Wang T, Huang XS, Li JY, Wang ZY. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem Int. 2013. https://doi.org/10.1016/j.neuint.2012.12.005.
Poggiali E, Cassinerio E, Zanaboni L, Cappellini MD. An update on iron chelation therapy. Blood Transfus. 2012. https://doi.org/10.2450/2012.0008-12.
Abbruzzese G, Cossu G, Balocco M, Marchese R, Murgia D, Melis M, Galanello R, Barella S, Matta G, Ruffinengo U, Bonuccelli U, Forni GL. A pilot trial of deferiprone for neurodegeneration with brain iron accumulation. Haematologica. 2011. https://doi.org/10.3324/haematol.2011.043018.
Rao SS, Portbury SD, Lago L, Bush AI, Adlard PA. The iron chelator deferiprone improves the phenotype in a mouse model of tauopathy. J Alzheimers Dis. 2020. https://doi.org/10.3233/JAD-200551.
Chand K, Rajeshwari R, Candeias E, Cardoso SM, Chaves S, Santos MA. Tacrine-deferiprone hybrids as multi-target-directed metal chelators against Alzheimer’s disease: a two-in-one drug. Metallomics. 2018. https://doi.org/10.1039/c8mt00143j.
Cappellini MD. Long-term efficacy and safety of deferasirox. Blood Rev. 2008. https://doi.org/10.1016/S0268-960X(08)70007-9.
Banerjee P, Sahoo A, Anand S, Bir A, Chakrabarti S. The oral iron chelator, deferasirox, reverses the age-dependent alterations in iron and amyloid-β homeostasis in rat brain: implications in the therapy of Alzheimer’s disease. J Alzheimers Dis. 2016. https://doi.org/10.3233/JAD-150514.
Kwan P, Ho A, Baum L. Effects of deferasirox in Alzheimer’s disease and tauopathy animal models. Biomolecules. 2022. https://doi.org/10.3390/biom12030365.
Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, Srinivas P, Sambamurti K, Rao KJ, Scancar J, Messori L, Zecca L, Zatta P. Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis. 2009. https://doi.org/10.3233/JAD-2009-1068.
Daniel S, Limson JL, Dairam A, Watkins GM, Daya S. Through metal binding, curcumin protects against lead- and cadmium-induced lipid peroxidation in rat brain homogenates and against lead-induced tissue damage in rat brain. J Inorg Biochem. 2004. https://doi.org/10.1016/j.**orgbio.2003.10.014.
Dairam A, Limson JL, Watkins GM, Antunes E, Daya S. Curcuminoids, curcumin, and demethoxycurcumin reduce lead-induced memory deficits in male Wistar rats. J Agric Food Chem. 2007. https://doi.org/10.1021/jf063446t.
Xu Y, Ku B, Cui L, Li X, Barish PA, Foster TC, Ogle WO. Curcumin reverses impaired hippocampal neurogenesis and increases serotonin receptor 1A mRNA and brain-derived neurotrophic factor expression in chronically stressed rats. Brain Res. 2007. https://doi.org/10.1016/j.brainres.2007.05.071.
Barik A, Mishra B, Shen L, Mohan H, Kadam RM, Dutta S, Zhang HY, Priyadarsini KI. Evaluation of a new copper(II)-curcumin complex as superoxide dismutase mimic and its free radical reactions. Free Radic Biol Med. 2005. https://doi.org/10.1016/j.freeradbiomed.2005.05.005.
Baum L, Ng A. Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J Alzheimers Dis. 2004. https://doi.org/10.3233/jad-2004-6403.
Yang H, Zeng F, Luo Y, Zheng C, Ran C, Yang J. Curcumin scaffold as a multifunctional tool for Alzheimer’s disease research. Molecules. 2022. https://doi.org/10.3390/molecules27123879.
Joseph JA, Shukitt-Hale B, Casadesus G. Reversing the deleterious effects of aging on neuronal communication and behavior: beneficial properties of fruit polyphenolic compounds. Am J Clin Nutr. 2005. https://doi.org/10.1093/ajcn/81.1.313S.
Babu PV, Liu D. Green tea catechins and cardiovascular health: an update. Curr Med Chem. 2008. https://doi.org/10.2174/092986708785132979.
Mandel S, Weinreb O, Amit T, Youdim MBH. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J Neurochem. 2004. https://doi.org/10.1046/j.1471-4159.2003.02291.x.
Xu YQ, Gao Y, Granato D. Effects of epigallocatechin gallate, epigallocatechin and epicatechin gallate on the chemical and cell-based antioxidant activity, sensory properties, and cytotoxicity of a catechin-free model beverage. Food Chem. 2021. https://doi.org/10.1016/j.foodchem.2020.128060.
Chen T, Yang Y, Zhu S, Lu Y, Zhu L, Wang Y, Wang X. Inhibition of Aβ aggregates in Alzheimer’s disease by epigallocatechin and epicatechin-3-gallate from green tea. Bioorg Chem. 2020. https://doi.org/10.1016/j.bioorg.2020.104382.
Mandel S, Amit T, Bar-Am O, Youdim MBH. Iron dysregulation in Alzheimer’s disease: multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog Neurobiol. 2007. https://doi.org/10.1016/j.pneurobio.2007.06.001.
Jahanshahi M, Khalili M, Margedari A. Naringin chelates excessive iron and prevents the formation of amyloid-beta plaques in the hippocampus of iron-overloaded mice. Front Pharmacol. 2021. https://doi.org/10.3389/fphar.2021.651156.
Tegoni M, Valensin D, Toso L, Remelli M. Copper chelators: chemical properties and bio-medical applications. Curr Med Chem. 2014. https://doi.org/10.2174/0929867321666140601161939.
Lovstad RA. A kinetic study on the copper-albumin catalyzed oxidation of ascorbate. Biometals. 2002. https://doi.org/10.1023/a:1020247323914.
Tardito S, Barilli A, Bassanetti I, Tegoni M, Bussolati O, Franchi-Gazzola R, Mucchino C, Marchiò L. Copper-dependent cytotoxicity of 8-hydroxyquinoline derivatives correlates with their hydrophobicity and does not require caspase activation. J Med Chem. 2012. https://doi.org/10.1021/jm301053a.
Mancino AM, Hindo SS, Kochi A, Lim MH. Effects of clioquinol on metal-triggered amyloid-β aggregation revisited. Inorg Chem. 2009. https://doi.org/10.1021/ic9014256.
White AR, Du T, Laughton KM, Volitakis I, Sharples RA, **linas ME, Hoke DE, Holsinger RMD, Evin G, Cherny RA, Hill AF, Barnham KJ, Li QX, Bush AI, Masters CL. Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem. 2006. https://doi.org/10.1074/jbc.M602487200.
Bareggi SR, Cornelli U. Clioquinol: review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci Ther. 2012. https://doi.org/10.1111/j.1755-5949.2010.00231.x.
Cahoon L. The curious case of clioquinol. Nat Med. 2009. https://doi.org/10.1038/nm0409-356.
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008. https://doi.org/10.1016/j.neuron.2008.06.018.
Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008. https://doi.org/10.1016/S1474-4422(08)70167-4.
Telpoukhovskaia MA, Orvig C. Werner coordination chemistry and neurodegeneration. Chem Soc Rev. 2013. https://doi.org/10.1039/C2CS35236B.
Bica L, Crouch PJ, Cappai R, White AR. Metallo-complex activation of neuroprotective signalling pathways as a therapeutic treatment for Alzheimer’s disease. Mol Biosyst. 2009. https://doi.org/10.1039/B816577G.
Treiber C, Simons A, Strauss M, Hafner M, Cappai R, Bayer TA, Multhaup G. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein in Alzheimer’s disease. J Biol Chem. 2004. https://doi.org/10.1074/jbc.M407410200.
Deraeve C, Pitié M, Mazarguil H, Meunier B. Bis-8-hydroxyquinoline ligands as potential anti-Alzheimer agents. New J Chem. 2007. https://doi.org/10.1039/B616085A.
Deraeve C, Boldron C, Maraval A, Mazarguil H, Gornitzka H, Vendier L, Pitié M, Meunier B. Preparation and study of new poly-8-hydroxyquinoline chelators for an anti-Alzheimer strategy. Chemistry. 2008. https://doi.org/10.1002/chem.200701024.
Deraeve C, Maraval A, Vendier L, Faugeroux V, Pitié M, Meunier B. Preparation of new bis(8-aminoquinoline) ligands and comparison with bis(8-hydroxyquinoline) ligands on their ability to chelate CuII and ZnII. Eur J Inorg Chem. 2008. https://doi.org/10.1002/ejic.200800924.
Cherny RA, Atwood CS, **linas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001. https://doi.org/10.1016/s0896-6273(01)00317-8.
Rodríguez-Rodríguez C, Telpoukhovskaia M, Orvig C. The art of building multifunctional metal-binding agents from basic molecular scaffolds for the potential application in neurodegenerative diseases. Coord Chem Rev. 2012. https://doi.org/10.1016/j.ccr.2012.03.008.
Scott LE, Orvig C. Medicinal inorganic chemistry approaches to passivation and removal of aberrant metal ions in disease. Chem Rev. 2009. https://doi.org/10.1021/cr9000176.
Frid P, Anisimov SV, Popovic N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res Rev. 2007. https://doi.org/10.1016/j.brainresrev.2006.08.001.
Reinke AA, Gestwicki JE. Insight into amyloid structure using chemical probes. Chem Biol Drug Des. 2011. https://doi.org/10.1111/j.1747-0285.2011.01110.x.
Hong MC, Kim YK, Choi JY, Yang SQ, Rhee H, Ryu YH, Choi TH, Cheon GJ, An GI, Kim HY, Kim Y, Kim DJ, Lee JS, Chang YT, Lee KC. Synthesis and evaluation of stilbene derivatives as a potential imaging agent of amyloid plaques. Bioorg Med Chem. 2010. https://doi.org/10.1016/j.bmc.2010.06.044.
Ono M, Haratake M, Mori H, Nakayama M. Novel chalcones as probes for in vivo imaging of beta-amyloid plaques in Alzheimer’s brains. Bioorg Med Chem. 2007. https://doi.org/10.1016/j.bmc.2007.07.052.
Newberg AB, Wintering NA, Plössl K, Hochold J, Stabin MG, Watson M, Skovronsky D, Clark CM, Kung MP, Kung HF. Safety, biodistribution and dosimetry of 123I-IMPY: a novel amyloid plaque-imaging agent for the diagnosis of Alzheimer’s disease. J Nucl Med. 2006;47:748–54.
Kung HF. The β-Amyloid Hypothesis in Alzheimer’s disease: seeing is believing. ACS Med Chem Lett. 2012. https://doi.org/10.1021/ml300058m.
Ono M, Watanabe H, Watanabe R, Haratake M, Nakayama M, Saji H. Diphenylpropynone derivatives as probes for imaging β-amyloid plaques in Alzheimer’s brains. Bioorg Med Chem Lett. 2011. https://doi.org/10.1016/j.bmcl.2010.11.058.
Pithadia AS, Kochi A, Soper MT, Beck MW, Liu Y, Lee S, DeToma AS, Ruotolo BT, Lim MH. Reactivity of diphenylpropyone derivatives toward metal-associated amyloid-β species. Inorg Chem. 2012. https://doi.org/10.1021/ic302084g.
Rodríguez-Rodríguez C, De Groot NS, Rimola A, Álvarez-Larena AN, Lloveras V, Vidal-Gancedo J, Ventura S, Vendrell J, Sodupe M, González-Duarte P. Design, selection, and characterization of thioflavin-based intercalation compounds with metal chelating properties for application in Alzheimer’s disease. J Am Chem Soc. 2009. https://doi.org/10.1021/ja806062g.
Geng J, Li M, Wu L, Ren J, Qu X. Liberation of copper from amyloid plaques: making a risk factor useful for Alzheimer’s disease treatment. J Med Chem. 2012. https://doi.org/10.1021/jm3003813.
Lee S, Zheng X, Krishnamoorthy J, Savelieff MG, Park HM, Brender JR, Kim JH, Derrick JS, Kochi A, Lee HJ, Kim C, Ramamoorthy A, Bowers MT, Lim MH. Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer’s disease. J Am Chem Soc. 2014. https://doi.org/10.1021/ja409801p.
Choi JS, Braymer JJ, Park SK, Mustafa S, Chae J, Lim MH. Synthesis and characterization of IMPY derivatives that regulate metal-induced amyloid-β aggregation. Metallomics. 2011. https://doi.org/10.1039/c0mt00077a.
Braymer JJ, Choi JS, DeToma AS, Wang C, Nam K, Kampf JW, Ramamoorthy A, Lim MH. Development of bifunctional stilbene derivatives for targeting and modulating metal-amyloid-β species. Inorg Chem. 2011. https://doi.org/10.1021/ic2012205.
Hindo SS, Mancino AM, Braymer JJ, Liu Y, Vivekanandan S, Ramamoorthy A, Lim MH. Small molecule modulators of copper-induced Aβ aggregation. J Am Chem Soc. 2009. https://doi.org/10.1021/ja907045h.
Choi JS, Braymer JJ, Nanga RPR, Ramamoorthy A, Lim MH. Design of small molecules that targer metal-A{beta} species and regulate metal-induced A{beta} aggregation and neurotoxicity. Proc Natl Acad Sci U S A. 2010. https://doi.org/10.1073/pnas.1006091107.
Jones MR, Service EL, Thompson JR, Wang MCP, Kimsey IJ, DeToma AS, Ramamoorthy A, Lim MH, Storr T. Dual-function triazole-pyridine derivatives as inhibitors of metal-induced amyloid-β aggregation. Metallomics. 2012. https://doi.org/10.1039/c2mt20113e.
Wahlström A, Hugonin L, Perálvarez-Marín A, Jarvet J, Gräslund A. Secondary structure conversions of Alzheimer’s Aβ(1–40) peptide induced by membrane-mimicking detergents. FEBS J. 2008. https://doi.org/10.1111/j.1742-4658.2008.06643.x.
Pithadia AS, Lim MH. Metal-associated amyloid-β species in Alzheimer’s disease. Curr Opin Chem Biol. 2012. https://doi.org/10.1016/j.cbpa.2012.01.016.
Zhang Y, Chen LY, Yin WX, Yin J, Zhang SB, Liu CL. The chelation targeting metal-Aβ40 aggregates may lead to formation of Aβ40 oligomers. Dalton Trans. 2011. https://doi.org/10.1039/C1DT00020A.
Sharma AK, Pavlova ST, Kim J, Finkelstein D, Hawco NJ, Rath NP, Kim J, Mirica LM. Bifunctional compounds for controlling metal-mediated aggregation of the aβ42 peptide. J Am Chem Soc. 2012. https://doi.org/10.1021/ja210588m.
Dedeoglu A, Cormier K, Payton S, Tseitlin KA, Kremsky JN, Lai L, Li X, Moir RD, Tanzi RE, Bush AI, Kowall NW, Rogers JT, Huang X. Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer’s amyloidogenesis. Exp Gerontol. 2004. https://doi.org/10.1016/j.exger.2004.08.016.
Scott LE, Telpoukhovskaia M, Rodríguez-Rodríguez C, Merkel M, Bowen ML, Page BDG, Grenn DE, Storr T, Thomas F, Allen DD, Lockman PR, Patrick BO, Adam MJ, Orvig C. N-Aryl substituted 3-(β-D-glucopyranosyloxy)-2-methyl-4(1H)-pyridinones as agents for Alzheimer’s therapy. Chem Sci. 2011. https://doi.org/10.1039/C0SC00544D.
Grasso GI, Arena G, Bellia F, Rizzarelli E, Vecchio G. Copper(II)-chelating homocarnosine glycoconjugate as a new multifunctional compound. J Inorg Biochem. 2014. https://doi.org/10.1016/j.**orgbio.2013.10.020.
Liu R, Barkhordarian H, Emadi S, Park CB, Sierks MR. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol Dis. 2005. https://doi.org/10.1016/j.nbd.2005.02.003.
Tan RX, Li WH, Pang JM, Zhong SM, Huang XY, Deng JZ, Zhou LY, Wu JQ, Wang XQ. Design, synthesis, and evaluation of 2,2′-bipyridyl derivatives as bifunctional agents against Alzheimer’s disease. Mol Divers. 2023. https://doi.org/10.1007/s11030-023-10651-5.
Zhao J, Shi Q, Tian H, Li Y, Liu Y, Xu Z, Robert A, Liu Q, Meunier B. TDMQ20, a specific copper chelator, reduces memory impairments in Alzheimer’s disease mouse models. ACS Chem Neurosci. 2021. https://doi.org/10.1021/acschemneuro.0c00621.
Pavlidis N, Kofinas A, Papanikolaou MG, Miras HN, Drouza C, Kalampounias AG, Kabanos TA, Konstandi M, Leondaritis G. Synthesis, characterization and pharmacological evaluation of quinoline derivatives and their complexes with copper(II) in in vitro cell models of Alzheimer’s disease. J Inorg Biochem. 2021. https://doi.org/10.1016/j.**orgbio.2021.111393.
Crnich E, Sanchez E, Havens MA, Kissel DS. Sulfur-bridging the gap: investigating the electrochemistry of novel copper chelating agents for Alzheimer’s disease applications. J Biol Inorg Chem. 2023. https://doi.org/10.1007/s00775-023-02013-1.
Radford RJ, Lippard SJ. Chelators for investigating zinc metalloneurochemistry. Curr Opin Chem Biol. 2013. https://doi.org/10.1016/j.cbpa.2013.01.009.
Colvin RA, Holmes WR, Fontaine CP, Maret W. Cytosolic zinc buffering and muffling: their role in intracellular zinc homeostasis. Metallomics. 2010. https://doi.org/10.1039/b926662c.
Sensi SL, Paoletti P, Koh JY, Aizenman E, Bush AI, Hershfinkel M. The neurophysiology and pathology of brain zinc. J Neurosci. 2011. https://doi.org/10.1523/JNEUROSCI.3454-11.2011.
Tóth K. Zinc in neurotransmission. Annu Rev Nutr. 2011. https://doi.org/10.1146/annurev-nutr-072610-145218.
Wilson M, Hogstrand C, Maret W. Picomolar concentrations of free zinc(II) ions regulate receptor protein-tyrosine phosphatase beta activity. J Biol Chem. 2012. https://doi.org/10.1074/jbc.C111.320796.
Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009. https://doi.org/10.1038/emboj.2009.338.
Huang YZ, Pan E, **ong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008. https://doi.org/10.1016/j.neuron.2007.11.026.
Huang YZ, McNamara JO. Neuroprotective effects of reactive oxygen species mediated by BDNF-independent activation of TrkB. J Neurosci. 2012. https://doi.org/10.1523/JNEUROSCI.0755-12.2012.
Veran J, Kumar J, Pinheiro PS, Athane A, Mayer ML, Perrais D, Mulle C. Zinc potentiates GluK3 glutamate receptor function by stabilizing the ligand binding domain dimer interface. Neuron. 2012. https://doi.org/10.1016/j.neuron.2012.08.027.
Wang T, Wang CY, Shan ZY, Teng WP, Wang ZY. Clioquinol reduces zinc accumulation in neuritic plaques and inhibits the amyloidogenic pathway in AbetaPP/PS1 transgenic mouse brain. J Alzheimers Dis. 2012. https://doi.org/10.3233/JAD-2011-111874.
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, Ingelsson M, Masters CL, Tanzi RE, Cummings JL, Herd CM, Bush AI. PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J Alzheimers Dis. 2010. https://doi.org/10.3233/JAD-2010-1390.
Corona C, Frazzini V, Silvestri E, Lattanzio R, La Sorda R, Piantelli M, Canzoniero LMT, Ciavardelli D, Rizzarelli E, Sensi SL. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS One. 2011. https://doi.org/10.1371/journal.pone.0017971.
Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, Sharples RA, Hill AF, Li QX, Masters CL, Barnham KJ, White AR. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J Biol Chem. 2008. https://doi.org/10.1074/jbc.M705957200.
Greenough MA, Volitakis I, Li QX, Laughton K, Evin G, Ho M, Dalziel AH, Camakaris J, Bush AI. Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J Biol Chem. 2011. https://doi.org/10.1074/jbc.M110.163964.
Pearson RG. Hard and soft acids and bases. J Am Chem Soc. 1963. https://doi.org/10.1021/ja00905a001.
Ambundo EA, Deydier MV, Ochrymowycz LA, Rorabacher DB. Kinetics and mechanism of copper(II) complex formation with tripodal aminopolythiaether and aminopolypyridyl ligands in aqueous solution. Inorg Chem. 2000. https://doi.org/10.1021/ic990904p.
Yang Y, Kawataki T, Fukui K, Koike T. Cellular Zn2+ chelators cause “dying-back” neurite degeneration associated with energy impairment. J Neurosci Res. 2007. https://doi.org/10.1002/jnr.21411.
Meeusen JW, Nowakowski A, Petering DH. Reaction of metal-binding ligands with the zinc proteome: zinc sensors and N, N, N′, N′-tetrakis(2-pyridylmethyl)ethylenediamine. Inorg Chem. 2012. https://doi.org/10.1021/ic2025236.
Lee JM, Kim YJ, Ra H, Kang SJ, Han S, Koh JY, Kim YH. The involvement of caspase-11 in TPEN-induced apoptosis. FEBS Lett. 2008. https://doi.org/10.1016/j.febslet.2008.04.056.
Carraway RE, Dobner PR. Zinc pyrithione induces ERK- and PKC-dependent necrosis distinct from TPEN-induced apoptosis in prostate cancer cells. Biochim Biophys Acta. 2012. https://doi.org/10.1016/j.bbamcr.2011.09.013.
Perez LR, Franz KJ. Minding metals: tailoring multifunctional chelating agents for neurodegenerative disease. Dalton Trans. 2010. https://doi.org/10.1039/b919237a.
Schugar H, Green DE, Bowen ML, Scott LE, Storr T, Bohmerle K, Thomas F, Allen DD, Lockman PR, Merkel M, Thompson KH, Orvig C. Combating Alzheimer’s disease with multifunctional molecules designed for metal passivation. Angew Chem Int Ed. 2007. https://doi.org/10.1002/anie.200603866.
Wu WH, Lei P, Liu Q, Hu J, Gunn AP, Chen MS, Rui YF, Su XY, **e ZP, Zhao YF, Bush AI, Li YM. Sequestration of copper from beta-amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J Biol Chem. 2008. https://doi.org/10.1074/jbc.M804722200.
Kay AR, Toth K. Is zinc a neuromodulator? Sci Signal. 2008. https://doi.org/10.1126/stke.119re3.
Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009. https://doi.org/10.1016/j.neuroscience.2008.01.061.
Kawabata E, Kikuchi K, Urano Y, Kojima H, Odani A, Nagano T. Design and synthesis of zinc-selective chelators for extracellular applications. J Am Chem Soc. 2005. https://doi.org/10.1021/ja044697q.
Zhang XA, Song D, Lippard SJ. A reversible pH-dependent intramolecular pyridine-aldehyde cyclization. J Org Chem. 2008. https://doi.org/10.1021/jo702394v.
Aalikhani M, Safdari Y, Jahanshahi M, Alikhani M, Khalili M. Comparison between hesperidin, coumarin and deferoxamine iron chelation and antioxidant activity against excessive iron in the iron overloaded mice. Front Neurosci. 2022. https://doi.org/10.3389/fnins.2021.811080.
Mazimba O. Umbelliferone: sources, chemistry and bioactivities review. Bull Fac Pharm Cairo Univ. 2017. https://doi.org/10.1016/j.bfopcu.2017.05.001.
Catapano MC, Karlíčková J, Tvrdý V, Sharma S, Prasad AK, Saso L, Chhillar AK, Kuneš J, Pour M, Parmar VS, Mladěnka P. Mono and dihydroxy coumarin derivatives: copper chelation and reduction ability. J Trace Elem Med Biol. 2018. https://doi.org/10.1016/j.jtemb.2017.11.014.
Singh R, Singh B, Singh S, Kumar N, Kumar S, Arora S. Umbelliferone—an antioxidant isolated from Acacia nilotica (L.) Willd. Ex. Del. Food Chem. 2010. https://doi.org/10.1016/j.foodchem.2009.11.022.
Mazimba O, Ma**da RRT, Modibedi C, Masesane IB, Cencic A, Chingwaru W. Tylosema esculentum extractives and their bioactivity. Bioorg Med Chem. 2011. https://doi.org/10.1016/j.bmc.2011.07.006.
Kanimozhi G, Prasad NR, Ramachandran S, Pugalendi KV. Umbelliferone modulates gamma-radiation induced reactive oxygen species generation and subsequent oxidative damage in human blood lymphocytes. Eur J Pharmacol. 2011. https://doi.org/10.1016/j.ejphar.2011.09.003.
Mladěnka P, Macáková K, Zatloukalová L, Reháková Z, Singh BK, Prasad AK, Parmar VS, Jahodar L, Hrdina R, Saso L. In vitro interactions of coumarins with iron. Biochimie. 2010. https://doi.org/10.1016/j.biochi.2010.03.025.
Kou X, Hu C, Pang Z, Zhang X, Wang H, Shen R, Yang A. A coumarin-based multifunctional chemosensor for Cu2+/Al3+ as an AD theranostic agent: synthesis, X-ray single crystal analysis and activity study. Anal Chim Acta. 2023. https://doi.org/10.1016/j.aca.2023.341818.
Wang H, Su M, Shi X, Li X, Zhang X, Yang A, Shen R. Design, synthesis, calculation and biological activity studies based on privileged coumarin derivatives as multifunctional anti-ad lead compound. Chem Biodivers. 2023. https://doi.org/10.1002/cbdv.202200867.
Du DX, Khang NHD, Tri NH, Nam PC, Thong NM. Exploring the multitarget activity of wedelolactone against Alzheimer’s disease: insights from in silico study. ACS Omega. 2023. https://doi.org/10.1021/acsomega.2c08014.
Zhang C, Yang K, Yu S, Su J, Yuan S, Han J, Chen Y, Gu J, Zhou T, Bai R, **e Y. Design, synthesis and biological evaluation of hydroxypyridinone-coumarin hybrids as multimodal monoamine oxidase B inhibitors and iron chelates against Alzheimer’s disease. Eur J Med Chem. 2019. https://doi.org/10.1016/j.ejmech.2019.07.031.
Hider RC, Roy S, Ma YM, Le Kong X, Preston J. The potential application of iron chelators for the treatment of neurodegenerative diseases. Metallomics. 2011. https://doi.org/10.1039/C0MT00087F.
Tripathi AC, Upadhyay S, Paliwal S, Saraf SK. Privileged scaffolds as MAO inhibitors: retrospect and prospects. Eur J Med Chem. 2018. https://doi.org/10.1016/j.ejmech.2018.01.003.
Carradori S, Silvestri R. New frontiers in selective human MAO-B inhibitors. J Med Chem. 2015. https://doi.org/10.1021/jm501690r.
Cobb CA, Cole MP. Oxidative and nitrative stress in neurodegeneration. Neurobiol Dis. 2015. https://doi.org/10.1016/j.nbd.2015.04.020.
Park JH, Ju YH, Choi JW, Song HJ, Jang BK, Woo J, Chun H, Kim HJ, Shin SJ, Yarishkin O, Jo S, Park M, Yeon SK, Kim S, Kim J, Nam MH, Londhe AM, Kim J, Cho SJ, Cho S, Lee C, Hwang SY, Kim SW, Oh SJ, Cho J, Pae AN, Lee CJ, Park KD. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci Adv. 2019. https://doi.org/10.1126/sciadv.aav0316.
Mi Z, Gan B, Yu S, Guo J, Zhang C, Jiang X, Zhou T, Su J, Bai R, **e Y. Dual-target anti-Alzheimer’s disease agents with both iron ion-chelating and monoamine oxidase-B inhibitory activity. J Enzyme Inhib Med Chem. 2019. https://doi.org/10.1080/14756366.2019.1634703.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic. Provided by the Grant Agency for Science of the Slovak Ministry of Education (Grant no. 1/0037/22).
Conflict of interest
The authors have no conflicts of interests to declare in relation to the contents of this review.
Availability of data and material
Not applicable.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for pulication
Not applicable.
Code availability
Not applicable.
Author contributions
Both authors have contributed equally to this work in terms of conceptualization, review of literature, writing, and draft revision. Both authors have also read and approved the final submitted manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gucký, A., Hamuľaková, S. Targeting Biometals in Alzheimer’s Disease with Metal Chelating Agents Including Coumarin Derivatives. CNS Drugs 38, 507–532 (2024). https://doi.org/10.1007/s40263-024-01093-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-024-01093-0