
Abstract
Background and Objective
Deficiencies of enzymes acting downstream of glucosylceramide synthase (GCS) can cause severe substrate accumulation. Venglustat is a small-molecule, brain-penetrant GCS inhibitor under investigation for multiple diseases involving pathogenic glycosphingolipid accumulation. Here, we evaluate the pharmacokinetics, safety, and tolerability of venglustat in healthy Chinese volunteers.
Methods
Study PKM16116 was a phase I, single-center, non-randomized, open-label study to investigate the pharmacokinetics, safety, and tolerability of a single 15 mg dose of orally administered venglustat in healthy Chinese volunteers aged 18 to 45 years.
Results
A total of 14 volunteers (7 male; 7 female) with a body mass index from 20.9 kg/m2 to 27.1 kg/m2 were enrolled. The median time to reach the venglustat maximum plasma concentration was 2.50 h post-dose. The mean terminal half-life of venglustat was 30.6 ± 7.40 h. The mean systemic exposures across all participants were 60.3 ± 17.3 ng/mL for the maximum plasma concentration, and 2280 ± 697 ng·h/mL for the area under the plasma concentration–time curve extrapolated to infinity. There were no relevant differences in venglustat pharmacokinetics between male and female volunteers. A post hoc cross-study comparison analysis showed comparable venglustat pharmacokinetics in Chinese and non-Chinese volunteers. Venglustat was safe and well tolerated in the current study (a total of five Grade 1 treatment-emergent adverse events were reported in three volunteers).
Conclusion
Venglustat showed a favorable pharmacokinetic, safety, and tolerability profile in healthy Chinese volunteers following a single oral 15 mg dose.
Clinical Trial Registry no.
CTR20201012 (http://www.chinadrugtrials.org.cn) registered on 24 February 2021 and ChiCTR2200066559 (http://www.chictr.org.cn) retrospectively registered on 9 December 2022.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A single oral dose of 15 mg venglustat in healthy Chinese volunteers demonstrated a favorable pharmacokinetic profile that is compatible with once daily oral dosing with no relevant differences between male and female participants. |
Pharmacokinetic parameters (Cmax and AUCinf) in healthy Chinese volunteers were generally comparable to those in non-Chinese volunteers in a post hoc cross-study comparison. |
Venglustat was safe and well tolerated in all Chinese volunteers, and there were no specific safety concerns identified. |
1 Introduction
Glycosphingolipids are plasma membrane components that are important for many cellular functions, including intracellular messaging, cell growth, and cell differentiation [1, 2]. In specific rare conditions, referred to as lysosomal storage diseases, pathogenic genetic deficiencies in intracellular metabolic pathway enzymes cause abnormalities in glycosphingolipid metabolism [1, 2]. Abnormal glycosphingolipid metabolism leads to lysosomal substrate accumulation, multisystemic impaired cell function, and subsequently, debilitating symptoms commonly seen in patients with lysosomal storage diseases.
Glucosylceramide synthase (GCS) is an enzyme involved in glycosphingolipid metabolism and is responsible for catalyzing the transfer of glucose to a ceramide lipid moiety to form glucosylceramide (GL-1) [3]. GL-1 is a key cellular structural building block for complex and higher-order glycosphingolipids, and is also important for cell function [3]. Inherited deficiencies of enzymes involved in substrate metabolism downstream of GCS can result in severe substrate accumulation as seen in lysosomal storage diseases such as Gaucher disease type 3 (GD3) [4], Fabry disease [5], and GM2 gangliosidoses [6].
One therapeutic method to improve the symptoms of substrate accumulation seen in these patients is via the inhibition of GCS and decreasing the formation of GL-1. By limiting the formation of GL-1, downstream pathogenic sphingolipid accumulation within the cell will be reduced. Venglustat is a novel, oral, small-molecule, GCS inhibitor which can cross the blood–brain barrier [7]. Venglustat is currently under investigation as a potential disease-modifying therapy for multiple lysosomal storage diseases, including GD3 [7], Fabry disease [8], and GM2 gangliosidoses [9]. Phase I studies conducted in healthy volunteers from the USA have demonstrated that venglustat has a favorable safety and tolerability profile [10]. Furthermore, the pharmacokinetic and dose-dependent pharmacodynamic profile of venglustat are compatible with once-daily oral dosing and consistent with the proposed mechanism of action (venglustat-mediated GCS inhibition) [10]. One year of 15 mg venglustat once daily in combination with a maintenance dose of imiglucerase enzyme replacement therapy showed acceptable safety and tolerability in patients with GD3 [11], and a confirmatory clinical study is currently ongoing to evaluate the efficacy and safety of venglustat treatment in patients with GD3 [12].
Approximately 80% of venglustat is metabolized by cytochrome P450 (CYP) 3A4 and eliminated renally, with a fraction of 26–33% excreted as parent compound. Although significant differences in the inter-individual variation in CYP3A4 activity have been reported (up to 202-fold), genetic polymorphism of CYP3A4 had very limited effects on metabolic activity [13]. Therefore, we hypothesize that there will be no ethnic differences in the clinical pharmacokinetics of venglustat among different racial populations. In this phase 1 study we aimed to assess the pharmacokinetic, safety, and tolerability of a single dose of 15 mg venglustat in healthy Chinese volunteers; to validate the hypothesis of pharmacokinetics similarities at this targeted dose between Chinese and non-Chinese healthy volunteers; and to support the clinical development of venglustat as a disease-modifying therapy for Chinese patients with lysosomal storage disease.
2 Methods
2.1 Study Design
Study PKM16116 was a phase I, single-center, non-randomized, open-label study to investigate the pharmacokinetics, safety, and tolerability of venglustat in healthy male and female Chinese volunteers and conducted between May 2021 and June 2021. The study consisted of three periods, with a total duration per volunteer of up to 31 days. The three periods were as follows: screening period, Day −21 to Day −1; observation period including one treatment day, Day 1 to Day 6; and follow-up period including an end-of-study visit at Day 8, Day 9, or Day 10.
Volunteers received a single 15 mg dose of orally administered venglustat hard capsule (venglustat malate, dose calculated with reference to free base) with 240 mL of tepid water and fasted for at least 10 h before and 4 h after dosing with venglustat.
The primary endpoint was to assess pharmacokinetic parameters of venglustat after a single-dose administration in healthy Chinese male and female volunteers (see Sect. 2.4 for more details). The secondary endpoint was to assess safety and tolerability of venglustat after a single-dose administration (see Sect. 2.5 for more details).
This study was registered at China Drug Trials Registry on 24 February 2021 (http://www.chinadrugtrials.org.cn, identifier: CTR20201012) and retrospectively registered at Chinese Clinical Trial Registry on 9 December 2022 (http://www.chictr.org.cn, identifier: ChiCTR2200066559), and conducted at a single study site, Bei**g Hospital, Bei**g, People’s Republic of China (approval no. 2020BJYYEC-176-02). The first volunteer was enrolled on 17 May 2021, and the last volunteer completed the study on 1 June 2021.
2.2 Study Population
Healthy Chinese volunteers aged between 18 years and 45 years, with a body weight of 55–100 kg if male or 50–90 kg if female, and a body mass index (BMI) value between 19.0 kg/m2 and 27.9 kg/m2 were enrolled. All volunteers were certified as healthy as assessed by a comprehensive clinical assessment, electrocardiogram (ECG), vital signs, and clinical laboratory assays. Men or women of childbearing potential were required to either practice true abstinence or use double contraceptive methods for the entire duration of the treatment.
Key exclusion criteria included: history or presence of clinically relevant cardiovascular, pulmonary, gastrointestinal, hepatic, renal, metabolic, hematological, neurological, osteomuscular, articular, psychiatric, systemic, ocular, or infectious disease (including hepatitis B or C, or human immunodeficiency virus 1 or 2), or signs of acute illness; frequent headaches and/or migraines, recurrent nausea and/or vomiting; blood donation (any volume) within 3 months of the study; symptomatic postural hypotension; presence or history of drug hypersensitivity, allergic disease or substance abuse; smoking (> 5 cigarettes/week equivalent, if unable to stop on-study); any medication (including St. John’s wort, herbal supplements, or Chinese traditional medicine) within 14 days before inclusion or within 5 times elimination or pharmacodynamic half-lives of the medication, whichever is longer; any vaccination within the last 28 days; citrus fruit or juice consumption within 5 days before or during the study. Women were not enrolled if they were pregnant or breastfeeding.
2.3 Sample Collection
The sample collection schedule was designed and optimized on the basis of the pharmacokinetic information from a previous clinical study [10]. Briefly, venous blood samples for pharmacokinetic measurements were collected before venglustat administration, and at 0.5 h, 1 h, 2 h, 3 h, 4 h, 5 h, 6 h, 8 h, 10 h, 12 h, 24 h, 48 h, 72 h, 96 h, and 120 h after venglustat administration. Venous blood samples of 2 mL were collected into K2-EDTA anticoagulant tubes and then centrifuged at 1300×g for 10 min at 4 °C to obtain the plasma fraction within 30 min of blood collection. Plasma samples were stored at −70 °C until analysis.
Venglustat concentrations in plasma were determined using a validated LC–MS/MS method with a lower limit of quantification (LLOQ) of 0.500 ng/mL. An Applied Biosystems Sciex API 5500 mass spectrometer, interfaced via an electrospray ionization probe with a Shimadzu high-performance liquid chromatography, was used.
The liquid chromatographic separation between venglustat and matrix components was achieved using an Agilent Zorbax Bonus-RP Column (2.1 × 50 mm, 1.8 μm particle size; column temperature maintained at 40 °C) using a mobile phase consisting of A, 0.1% formic acid in water (v/v), and B, 0.1% formic acid in acetonitrile (v/v). The gradient started at 95% A, 5% B, ram** to 15% B at 5 min, 95% B at 5.1–6 min, then back to 5% B at 6.1 min and stopped at 7 min. The flow rate was 0.8 mL/min.
Mass spectroscopy used positive ion mode with ionspray voltage of 5500 eV; source temperature at 600 °C; both gas 1 and gas 2 at 55 psi N2; curtain gas at 40 psi; and a collisionally activated dissociation of 9. The acquisition duration was 7 min with multiple reaction monitoring modes, that is, monitoring m/z ratios of 390.2 → 220.0 for venglustat and 396.2 → 226.0 for deuterated internal standard. The lower limit of quantification (LLOQ) was 0.500 ng/mL and the quantitation range was 0.500 ng/mL to 500 ng/mL using 20 μL plasma sample and a 6.00 μL injection volume. Intra-batch precision maximum coefficient of variation (CV) was 13.8%, and intra-batch accuracy ranged from −11.2 to −8.4%. Inter-batch in LLOQ precision was 8.8% CV and accuracy was −10.0%.
2.4 Primary Outcomes: Pharmacokinetic Assessments
Pharmacokinetic parameters were determined using Phoenix WinNonlin Professional software (version 8.1, Certara, Inc., Princeton, New Jersey). The maximum plasma concentration (Cmax), the time taken to reach Cmax (Tmax), and the time of last measurable concentration (tlast) were obtained from the observed data. The area under the plasma concentration–time curve from time zero to the last measurable concentration (AUClast) was calculated using the trapezoidal method and that from zero to infinity (AUCinf) was generated according to the equation of:
where Clast is the last measurable concentration and λz is the slope of the regression line for the terminal phase of the plasma concentration–time curve in semi-logarithmic scale. The terminal half-life (t1/2z) was calculated using the relationship 0.693/λz. The apparent systemic clearance from plasma (CL/F) was estimated by dividing the oral dose by AUCinf. The apparent volume of distribution at steady state (Vss/F) was calculated by multiplying the CL/F by the mean residence time (MRT). MRT is the mean time the drug remains in the body and was estimated by dividing the AUCinf by AUMC, where AUMC is the area under the curve of moments calculated using the trapezoidal method.
2.5 Secondary Outcomes: Safety Analysis
Safety was evaluated by assessing treatment-emergent adverse events (TEAEs) throughout the study and by using ECG parameters, clinical laboratory evaluations, and vital signs. TEAEs are defined as the adverse events that developed or worsened during the time period from venglustat administration up to the last study visit. Adverse events were graded according to National Cancer Institute-Common Terminology Criteria for Adverse Events v5.0 [14].
2.6 Statistical Analyses
Pharmacokinetic parameters were calculated and summarized using descriptive statistics by Phoenix WinNonlin software. Safety analysis (including ECG parameters, clinical laboratory evaluations, and vital signs) were based on the review of individual values and descriptive statistics. The statistical analyses were conducted using SAS (Statistical Analysis Software, Cary, North Carolina).
3 Results
3.1 Baseline Demographics
The study enrolled 14 volunteers balanced by gender (7 male; 7 female). All volunteers completed the study. Volunteers were aged between 22 years and 44 years (mean ± standard deviation [SD] 32.4 ± 7.6 years). The mean ± SD baseline weight was 64.9 ± 11.4 kg, and the mean ± SD baseline BMI was 23.6 ± 1.72 kg/m2.
3.2 Primary Outcomes: Pharmacokinetics
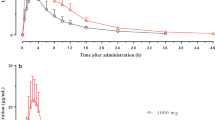
With a single oral dose of 15 mg, venglustat was absorbed with a median tmax of 2.50 h post-dose across all volunteers (Table 1). Median tmax occurred slightly later in female volunteers at 3.00 h post-dose compared with 2.00 h post-dose in males. After reaching Cmax, venglustat was then eliminated with mean t1/2z of 30.6 h across all volunteers. In female volunteers, t1/2z was shorter with a mean of 24.6 h versus 36.6 h in male volunteers. Total systemic exposures based on mean AUClast and AUCinf were 2110 ng·h/mL and 2280 ng·h/mL, respectively (Fig. 1a). Systemic exposures within the first 24 h following dosing were similar between female and male volunteers. Mean AUClast and AUCinf were approximately 12.4% and 19.4% lower, respectively, in female volunteers compared with male volunteers (Fig. 1b).

Plasma concentration–time profiles (mean ± SD) following single-dose oral administration of 15 mg venglustat. (a) overall ; (b) by gender. h, hour; SD, standard deviation
In a post hoc analysis, venglustat pharmacokinetic parameters were compared between healthy Chinese volunteers in the current study (study PKM16116 [CTR20201012]) and healthy non-Chinese volunteers (data from phase I studies: TDU12766 [N = 6; NCT01674036] [10] and BEQ15920 [unpublished; N = 33; NCT02906020]; Fig. 2). On the basis of the linear pharmacokinetic property of venglustat demonstrated in study TDU12766 (single-dose escalation study), data at a dose level of 15 mg venglustat l-malate from TDU12766 were normalized to 15 mg venglustat free base to match the dose level for comparison.

Dose normalized individual and mean ± SD pharmacokinetic parameters in healthy Chinese volunteers (study PKM16116 [ChiCTR2200066559]) and non-Chinese volunteers (phase I studies TDU12766 [NCT01674036] [10] and unpublished BEQ15920 [NCT02906020]). (a) Cmax; (b) AUCinf. AUCinf, area under the plasma concentration–time curve extrapolated to infinity; Cmax, maximum plasma concentration; SD, standard deviation
The mean ± SD Cmax was 60.3 ± 17.3 ng/mL, 70.9 ± 22.4 ng/mL, and 53.3 ± 11.1 ng/mL for studies PKM16116, TDU12766, and BEQ15920 (15 mg venglustat bioequivalence study), respectively. The mean ± SD AUCinf was 2280 ± 697 ng·h/mL, 2765 ± 803 ng·h/mL, and 2110 ± 545 ng·h/mL, and the mean ± SD t1/2z was 30.6 ± 7.40 h, 29.7 ± 7.12 h, and 32.7 ± 6.91 h for studies PKM16116, TDU12766, and BEQ15920, respectively.
3.3 Secondary Outcomes: Safety and Tolerability
There were no deaths, treatment-emergent serious adverse events, TEAEs leading to permanent study discontinuation, or any Grade ≥ 3 TEAEs reported. Only 3 out of 14 volunteers (21.4%) experienced at least one TEAE (total of five TEAEs reported) during the study; there were five events in total under the primary system organ class of investigations (the most frequently reported was the presence of white blood cells in urine; 2/14 [14.3%]) and no events under the primary system organ class of psychiatric disorders or nervous system disorders were reported in this phase I study of a brain-penetrant GCS inhibitor. All TEAEs reported during the study were of Grade 1 intensity and recovered without any corrective treatment.
Only one volunteer experienced a transient increase in alanine aminotransferase levels (2.08 × upper limit of normal) without any signs or symptoms. The volunteer recovered from the event 5 days later without any corrective treatment.
No clinically meaningful trends were observed for hematological or biochemical values in the study (Table 2). There were no potentially clinically significant abnormalities for metabolic function, electrolytes, renal function, or liver function observed during the study. There were no clinically meaningful findings in the vital sign measurements or ECG assessments related to safety.
4 Discussion
This is the first study conducted in Chinese healthy volunteers to assess the pharmacokinetics, safety, and tolerability of oral administration of venglustat. The results also help to provide further confirmation in the similarities of the properties of venglustat across different ethnic populations. In the healthy Chinese volunteers who took part in this study, there were no specific safety concerns associated with oral administration of venglustat, thus supporting a favorable safety and tolerability profile. Although there were slight differences in venglustat pharmacokinetics between genders, these were not considered to be clinically relevant and should be interpreted with caution due to the small sample size for each gender.
In previous single-center first-in-human phase I (TDU12766: venglustat l-malate 2–150 mg equivalent to 1.5–112 mg free venglustat [NCT01674036]) and single-dose food-effect studies (FED12767: 5 mg l-malate venglustat equivalent to 3.7 mg free venglustat [NCT01674036]), venglustat demonstrated linear pharmacokinetics, rapid absorption, systemic exposure unaffected by food, and low apparent systemic clearance in healthy volunteers from the USA [10]. Although ethnic factors may influence the pharmacokinetics, efficacy, and safety of a drug across different populations [15], no ethnic differences were anticipated for venglustat pharmacokinetics due to its evidenced in vitro and in vivo properties, including linear pharmacokinetics, negligible food effect [10], moderate human plasma protein binding, and the predominant role of CYP3A4 in metabolism (unpublished data).
Post hoc analysis of data from this study, the first-in-human phase I study (TDU12766), and another phase I study (BEQ15920) identified that the pharmacokinetic parameters Cmax and AUCinf of venglustat in healthy Chinese and non-Chinese volunteers were generally comparable (Fig. 2). These results suggest that the systemic exposures of venglustat are similar in healthy Chinese and non-Chinese volunteers.
A phase II study by Peterschmitt et al. of low, mid, and high doses (4 mg, 8 mg, and 15 mg) of venglustat in Japanese (N = 9) and non-Japanese (N = 13) participants with Parkinson’s disease associated to a glucocerebrosidase gene mutation showed comparable pharmacokinetics of venglustat in ethnically different Japanese and non-Japanese patients [16]. The study reported dose-proportional increases in venglustat exposure in both plasma and cerebrospinal fluid (CSF) over 4 weeks in both participant populations. At Week 4 following once-daily administration, venglustat was absorbed in plasma with a median tmax of 2.1–4.3 h in Japanese participants, and 2.0–3.6 h in non-Japanese participants, generally comparable to the tmax data in the current study (2.5 h). At Week 4, the mean Cmax was 56–145 ng/mL versus 42–136 ng/mL, and the mean AUC0–24 was 1130–3150 ng·h/mL versus 766–2510 ng·h/mL in Japanese versus non-Japanese participants, respectively.
A possible limitation of our study is that CSF was not collected from the healthy volunteers for pharmacokinetic and biomarker assessment. As such, the study was unable to assess observations relating to drug penetration and pharmacology characterization. It is important to note that venglustat exposure in CSF was assessed in patients with Parkinson’s disease and a glucocerebrosidase gene mutation in the Peterschmitt et al. study [16], and was generally comparable in Japanese (mean CSF; 2–10 ng/mL) and non-Japanese participants (mean CSF; 2–6 ng/mL) [16]. As there is little cross-ethnic sensitivity in venglustat pharmacokinetics, we assume that venglustat exposure in the CSF of Chinese participants may have a similar range to that seen in non-Chinese participants. Nevertheless, assessment of venglustat exposure in CSF is expected to be evaluated in Chinese and other non-Chinese participants in ongoing multi-regional clinical trials. Another limitation of this study is that only a single dose of venglustat with the targeted therapeutic dose of 15 mg was administered, meaning we were unable to establish the overall effect on pharmacodynamic endpoints such as plasma GL-1 and monosialodihexosylganglioside (GM3). For this pharmacodynamic assessment of venglustat in heathy volunteers, repeat dosing of venglustat would be required. Although only a single dose of 15 mg venglustat was investigated in this study, data from global first-in-human [10], unpublished data from in vitro drug metabolism and pharmacokinetic, and other earlier phase I clinical pharmacological studies, indicate no time-dependent pharmacokinetics for venglustat. Considering the lack of ethnic sensitivity in pharmacokinetics from the current and previous studies [10, 16], time-dependency differences are not expected in the volunteers in the current studies, and therefore, single-dose pharmacokinetic data can predict the repeat-dose pharmacokinetics. Furthermore, pharmacokinetic, pharmacodynamic, and safety data from Chinese patients are being collected in ongoing multi-regional clinical trials, which will help broaden our understanding of the properties of venglustat in this population.
5 Conclusions
The findings of this study demonstrate that a single oral administration of 15 mg venglustat has a favorable pharmacokinetic profile, comparable to data from non-Chinese volunteers. Furthermore, it crucially demonstrates that venglustat has a favorable safety and tolerability profile within healthy Chinese volunteers. Overall, these findings support the global clinical development of venglustat as an oral GCS inhibitor for treatment of lysosomal storage diseases in the Chinese population.
References
Shayman JA. Targeting glucosylceramide synthesis in the treatment of rare and common renal disease. Semin Nephrol. 2018;38(2):183–92.
Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19(3):175–91.
Bleicher RJ, Cabot MC. Glucosylceramide synthase and apoptosis. Biochim Biophys Acta. 2002;1585(2–3):172–8.
Daykin EC, Ryan E, Sidransky E. Diagnosing neuronopathic Gaucher disease: new considerations and challenges in assigning Gaucher phenotypes. Mol Genet Metab. 2021;132(2):49–58.
Welford RWD, Muhlemann A, Garzotti M, Rickert V, Groenen PMA, Morand O, et al. Glucosylceramide synthase inhibition with lucerastat lowers globotriaosylceramide and lysosome staining in cultured fibroblasts from Fabry patients with different mutation types. Hum Mol Genet. 2018;27(19):3392–403.
Leal AF, Benincore-Florez E, Solano-Galarza D, Garzon Jaramillo RG, Echeverri-Pena OY, Suarez DA, et al. GM2 Gangliosidoses: clinical features, pathophysiological aspects, and current therapies. Int J Mol Sci. 2020;21(17):6213.
Marshall J, Sun Y, Bangari DS, Budman E, Park H, Nietupski JB, et al. CNS-accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic gaucher disease. Mol Ther. 2016;24(6):1019–29.
Ashe KM, Budman E, Bangari DS, Siegel CS, Nietupski JB, Wang B, et al. Efficacy of enzyme and substrate reduction therapy with a novel antagonist of glucosylceramide synthase for Fabry disease. Mol Med. 2015;21:389–99.
A multinational, randomized, double-blind, placebo-controlled study to assess the efficacy, pharmacodynamics, pharmacokinetics, and safety of venglustat in late-onset GM2 (AMETHIST). https://ClinicalTrials.gov/show/NCT04221451. Accessed 9 Jan 2020.
Peterschmitt MJ, Crawford NPS, Gaemers SJM, Ji AJ, Sharma J, Pham TT. Pharmacokinetics, pharmacodynamics, safety, and tolerability of oral venglustat in healthy volunteers. Clin Pharmacol Drug Dev. 2021;10(1):86–98.
Schiffmann R, Cox TM, Dedieu JF, Gaemers SJM, Hennermann JB, Ida H, et al. Venglustat combined with imiglucerase for neurological disease in adults with Gaucher disease type 3: the LEAP trial. Brain. 2023;146(2):461–74.
Study to evaluate the efficacy and safety of venglustat in adult and pediatric patients with Gaucher disease type 3 (LEAP2MONO). https://clinicaltrials.gov/ct2/show/NCT05222906. Accessed 3 Feb 2022.
Gao N, Tian X, Fang Y, Zhou J, Zhang H, Wen Q, et al. Gene polymorphisms and contents of cytochrome P450s have only limited effects on metabolic activities in human liver microsomes. Eur J Pharm Sci. 2016;92:86–97.
Freites-Martinez A, Santana N, Arias-Santiago S, Viera A. Using the Common Terminology Criteria for Adverse Events (CTCAE—Version 5.0) to evaluate the severity of adverse events of anticancer therapies. Actas Dermosifiliogr (Engl Ed). 2021;112(1):90–2.
International Conference on Harmonisation; guidance on ethnic factors in the acceptability of foreign clinical data; availability--FDA. Notice. Fed Regist. 1998;63(111):31790–6.
Peterschmitt MJ, Saiki H, Hatano T, Gasser T, Isaacson SH, Gaemers SJM, et al. Safety, pharmacokinetics, and pharmacodynamics of oral venglustat in patients with Parkinson’s disease and a GBA mutation: results from part 1 of the randomized, double-blinded, placebo-controlled MOVES-PD trial. J Parkinsons Dis. 2022;12(2):557–70.
Acknowledgements
The authors acknowledge Yongzhen Gu and Nancy Tang for study conduct; Stefanie Liang for development of the clinical study report; Irene Zang for coordinating the biological sample analysis; Jyoti Sharma for the pharmacokinetic data discussion; and all the volunteers who participated in this study. Editorial support in the preparation of this publication was provided by Nikita Vekaria, PhD, of Elevate Medical Affairs (a division of Envision Pharma Group), contracted by Sanofi for publication support services.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Sanofi.
Conflict of interest
Yingxin Li, Li Li, and Kong **n are employees of Sanofi and may hold shares and/or stock options (< 5%) in the company. Yang Li, Wei Xue, Titi Wang, and Aixin Shi have no conflicts of interest.
Data availability
The original data used for analysis in the current study will not be openly shared due to confidentiality concerns.
Code availability
Not applicable.
Author contributions
YL, LL, and AS were responsible for study concept and study design. YL, YL, LL, WX, TW, and AS were responsible for study conduct and data acquisition. YL, LL, and AS were responsible for analysis and interpretation. YL, YL, LL, WX, KX, TW, and AS were responsible for the manuscript (draft, review, and approval). The authors, individually and collectively, are responsible for all content and editorial decisions and received no payment from Sanofi directly or indirectly (through a third party) related to the development/presentation of this publication.
Ethics approval
The study was approved with an approval number of 2020BJYYEC-176-02 by the Ethics Committee of Bei**g Hospital. The study was undertaken in accordance with the consensus ethics principles derived from international ethics guidelines, including the Declaration of Helsinki, and the International Council of Harmonization guidelines for Good Clinical Practice. All volunteers gave their written informed consent prior to any study-related procedure.
Consent to participate
All volunteers gave their written informed consent prior to any study-related procedure.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Li, Y., Li, Y., Li, L. et al. Pharmacokinetics, Safety, and Tolerability of Single-Dose Orally Administered Venglustat in Healthy Chinese Volunteers. Clin Drug Investig 43, 413–420 (2023). https://doi.org/10.1007/s40261-023-01275-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-023-01275-6