
Abstract
Introduction
Kukoamine B mesylate (KB) is a mesylate chrysamine B targeting lipopolysaccharides and CpG DNA, two potential treatment targets in sepsis.
Methods
This first-in-human, randomized, double-blind, placebo-controlled, phase I study was conducted from July 2014 to May 2015 to explore the safety, tolerability, and pharmacokinetics of KB in healthy subjects. This study consisted of a pre-phase (four participants; KB at 0.005 mg/kg) and a dose escalation phase (eight participants/dose group, randomized 6:2 to KB or placebo; KB at 0.02, 0.04, 0.08, 0.12, 0.24, and 0.48 mg/kg). The primary endpoint was safety.
Results
Fifty-two participants were enrolled, including four in the pre-phase and 48 in the dose escalation phase. Among the 40 participants who received KB, 12 (30.0%) experienced adverse events (AEs), while two (16.7%) experienced AEs among 12 participants who received the placebo. The most common AEs in the KB group were headache (5.0%), influenza (5.0%) and positive white blood cell in urine (5.0%). After the administration of KB, the mean plasma elimination half was around 1.61–4.24 h. The relationship between the KB plasma exposure of KB and the administered dose was not linear. The percentage of cumulative urinary excretion of KB was similar among the different dose groups (21.7–35.2%) and the urinary excretion of KB decreased significantly about 8 h after administration.
Conclusions
Single-dose KB demonstrated favorable safety and tolerability in healthy subjects at the dose level of 0.005–0.48 mg/kg. KB exhibited a non-linear pharmacokinetic profile with a half-life of about 1.61–4.24 h, which mainly distributed in plasma.
Trial Registration
ClinicalTrials.gov identifier, NCT02219971.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
This first-in-human, randomized, double-blind, placebo-controlled, phase I study was conducted to explore the safety, tolerability, and pharmacokinetics of KB in healthy subjects. |
What was learned from this study? |
Single-dose KB demonstrated favorable safety and tolerability in healthy subjects at the dose level of 0.005–0.48 mg/kg. |
The findings of this study provide preliminary evidence for further clinical trials verifying the efficacy and safety of KB in sepsis. |
Introduction
Sepsis is a life-threatening organ dysfunction caused by the dysregulated host response to infection [1, 2]. Approximately 750,000 cases of sepsis occur annually in the United States [3, 4]. In China, the incidence of sepsis is 20.6 cases per 100 intensive care unit (ICU) admissions, with a 30-day mortality of 36% [Endpoints The primary endpoint was safety, which included AEs, clinical laboratory indicators, electrocardiogram, and vital signs. The severity of the AEs was determined as mild (did not affect daily life, no need for medical intervention), moderate (affected daily life, might require medical intervention), and severe (significantly affected daily life, required medical intervention). It was up to the investigators to determine whether the AEs were drug-related. The secondary endpoints were the pharmacokinetic indicators: time-to-maximum (Tmax), peak concentration (Cmax), areas under the curve (AUC0-t and AUC0-∞), clearance (CL), distribution volume (Vd), half-life (T1/2), prototype drug, and metabolites in urine and fecal samples. In the pre-phase, blood samples were collected before administration and after 20 and 40 min of infusion, immediately after the end of the infusion, 10, 20, 30, and 45 min, and 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, and 24 h after the end of infusion. Urine samples were collected before administration and 0–4, 4–8, 8–12, 12–24, and 24–48 h after the start of administration. In the dose escalation phase, the sampling points were adjusted according to the blood drug-concentration results in the pre-phase: before administration, 20 and 40 min after the start of the infusion, immediately after the end of infusion, and 30 min and 1, 2, 3, 4, 6, 8, 12, 16, and 24 h after the end of infusion. The sampling points of urine samples were adjusted as follows: before administration and 0–5, 5–9, 9–13, 13–25, and 25–49 h after the end of infusion. The content of KB and metabolites were determined quantitatively by liquid chromatography-mass spectrometry using the urine samples from participants received 0.12, 0.24, and 0.48 mg/kg KB. The participants in the 0.04, 0.08, 0.24, and 0.48 mg/kg dose groups collected fecal samples from the day before to 48 h after administration. In the context of this phase I clinical trial, the primary focus was on assessing the safety, tolerability, and pharmacokinetics of KB. Consequently, sample size calculations aimed at detecting specific treatment effects were not performed. The pharmacokinetic parameters were computed using non-compartmental analyses and Phoenix WinNonlin 6.3 (Pharsight). The linear correlations between the pharmacokinetic parameters (AUC0-inf, AUClast, and Cmax) of single administration and the doses were evaluated using Power Model in SAS 9.3 (SAS Institute Inc. Cary, NC, USA) using PROC MIXED (confidence level of 0.1). For AUC, the acceptable criteria for 90% confidence interval were 0.954–1.046 for AUC and 0.941–1.058 for Cmax. The continuous data that conformed to the normal distribution were presented as means ± standard deviation, and those that did not conform to the normal distribution were presented as median (range). The categorical data were presented as n (%). All results in this study were presented using descriptive statistics.Statistical Analysis
Results
Baseline Characteristics of the Participants
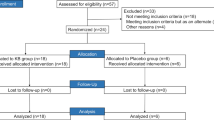
Fifty-two healthy participants were included in this study, including four in the pre-phase and 48 in the dose escalation phase (Fig. 1). In the dose escalation phase, 36 subjects (six in each dose group) received KB, and 12 received the placebo. Supplementary Table S1 presents the baseline characteristics of the participants.

Flow chart
Safety and Tolerability
All participants completed the KB or placebo dosing according to the study protocol. In the total population, 14 participants (26.9%) experienced AEs. Among the participants who received KB, 12 (30.0%) experienced AEs. Among those who received the placebo, two (16.7%) experienced AEs (Table 1). The common AEs in the KB group were headache (5.0%), influenza (5.0%), and positive white blood cell in urine (5.0%).
All AEs were mild except for one case of serum creatine phosphokinase elevation, which was judged to be moderate. All AEs recovered spontaneously without any treatment. No serious AEs occurred. Except for two cases of influenza, which were judged by the investigators as possibly unrelated to the study drug, all other AEs were judged as possibly related to the study drug. No participants stopped the study because of AEs.
Pharmacokinetics
The detailed pharmacokinetic parameters are shown in Table 2. After the administration of 0.005, 0.02, 0.04, 0.08, 0.12, 0.24, and 0.48 mg/kg of KB, the plasma elimination half-lives were 1.61 ± 0.14, 2.25 ± 0.44, 2.78 ± 0.41, 3.97 ± 0.56, 3.20 ± 0.87, 4.24 ± 0.75, and 3.75 ± 0.34 h, respectively.
After a single intravenous infusion of 0.005, 0.02, 0.04, 0.08, 0.12, 0.24, and 0.48 mg/kg of KB in 40 healthy subjects, the plasma concentrations of KB increased gradually after the start of intravenous infusion and decreased rapidly after the end of infusion. The plasma drug–time curves of KB in each dose group were similar in shape (Fig. 2A). The increase of KB exposure that increased with the administered dose was slightly higher than that of the dose increase, and no dose-linear relationship was observed.

A Mean time–concentration curves. B Mean urine cumulative excretion curves
The evaluation of the linear relationship using the Power model showed that the plasma exposure of KB (AUC and Cmax, as well as body weight-adjusted AUC and Cmax) increased with increasing administered dose, but the proportion of increase was slightly higher than that of the dose increase, approximately 10–15% above the proportion, a strict dose-linear relationship was not shown, and neither the point estimate nor its 90% confidence interval fell within the acceptable range (Table 3). As shown in Fig. 3, after the intravenous infusion of KB at doses ranging from 0.005 to 0.48 mg/kg, the incidence of AEs was independent of dose and systemic exposure.

Relationships between adverse event rates and kukoamine B mesylate exposure (A) and maximal concentration (B)
Compared with the mean plasma drug concentrations of KB in the other dose groups, the drug concentrations of KB in whole blood were lower in the 0.24 and 0.48 mg/kg-dose groups, and the ratio of whole blood/plasma drug concentrations (B/P ratios) were 0.43 and 0.47, respectively, suggesting that KB was mainly distributed in the plasma. The whole blood T1/2 of KB was 1.59 ± 0.29 and 1.78 ± 0.19 h, respectively, which were lower than the plasma T1/2 (Supplementary Table S2). The whole blood concentration–time curves of KB at 0.24 and 0.48 mg/kg showed that the whole blood concentrations of KB gradually increased after the start of intravenous infusion and decreased rapidly after stop** the infusion. The whole blood concentration–time curves of KB were similar in shape for the two dose groups, with a gradual increase in whole blood exposure with increasing doses (Supplementary Figure S1).
The cumulative urinary excretion of KB gradually increased with the increased administered dose, and the percentage of cumulative urinary excretion of each dose group was similar, ranging from 21.7% to 35.2% (Fig. 2B). About 8 h after administration, the excretion of the prototype KB in the urine was significantly reduced. In quantitative analysis of KB and metabolite in urine samples, the average percentage of KB prototypes in urine samples during 49 h after KB administration in relation to the administrated KB was 39.65%, 33.97%, and 26.29% in 0.12, 0.24 and 0.48 mg/kg-dose group, respectively. The highest level of metabolite in urine sample was M7 (bis-methylated product), which accounted for 8.07%, 9.98%, and 9.88% of the dose administered in 0.12, 0.24, and 0.48 mg/kg-dose group, respectively, followed by M11 (bis-methylated, oxidatively deaminated carboxylic acid derivative, accounted for 4.01%, 5.75%, and 5.28%). The content of the other two forms of metabolites (M1, methylated N-desalkyl product; M4, cysteine binding product) were too little to be quantitatively analyzed (Supplementary Figure S2).
Discussion
This first-in-human phase I trial aimed to explore the safety, tolerability, and pharmacokinetics of KB in healthy subjects. The results suggest that a single dose of KB demonstrated favorable safety and tolerability in healthy subjects at the dose level of 0.005–0.48 mg/kg. KB exhibited a non-linear pharmacokinetic profile with a half-life of about 1.61–4.24 h and was mainly distributed in plasma. The findings of this study provide preliminary evidence for further clinical trials verifying the efficacy and safety of KB in sepsis.
In this study, all AEs were mild except for one case of moderately elevated creatinine kinase. The most common AEs were influenza (5%), which was ruled to be possibly unrelated to KB, and headaches and positive white blood cell in urine, which were considered possibly related. A previous study in high-fat-fed rats showed that KB could decrease the levels of blood lipids and inflammatory cytokines and improve the markers of oxidative stress, but there were no data about common safety parameters like liver and kidney functions [12]. Another study revealed a decrease in inflammatory parameters in mice exposed to LPS without apparent toxicity [10]. Although these results are limited and preliminary, they suggest the safety of KB in humans.
Based on the present preliminary pharmacokinetic data, the exposure of KB in healthy subjects increased with the increase of the administered dose, but the proportion of the increase was slightly higher than that of the dose increase, which might be related to the characteristics of the study drug and factors such as the limited number of subjects. Still, such a non-linear relationship was also observed in a simulation study [13]. The whole blood T1/2 of KB was lower than the plasma T1/2, possibly because the lower limit of quantification (LLOQ) of the whole blood-based detection method was 100 times the LLOQ of the plasma-based detecting method, which made the whole blood sample at the end of the elimination phase less detectable than the plasma sample, resulting in different T1/2 values. A modeling study proposed a simulation model for calculating the optimal dose of KB in patients with sepsis [13], but these results are theoretical at best and actual observations remain the best course. Another simulation study proposed that single dosing of KB would be recommended for clinical trials [14].
This study has limitations. The pharmacokinetic data collected represented the best case in healthy subjects and did not include variability due to patient covariates. Due to the small sample size, the safety profile needs to be further confirmed.
Conclusions
In this randomized, double-blind, placebo-controlled, single-dose phase I trial, single-dose KB demonstrated favorable safety and tolerability in healthy subjects at the dose level of 0.005–0.48 mg/kg. KB exhibited a non-linear pharmacokinetic profile with a half-life of about 1.61–4.24 h and was mainly distributed in plasma. The urinary excretion of KB decreased significantly about 8 h after administration. The main metabolic pathways of KB were bimethylation, carbonylation, N-desalkylation and cysteamine binding. The safety and efficacy of KB will be further investigated in patients with sepsis.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
References
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–10.
Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021;47:1181–247.
Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. The Lancet. 2020;395:200–11.
Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther. 2012;10:701–6.
**e J, Wang H, Kang Y, Zhou L, Liu Z, Qin B, et al. The epidemiology of sepsis in Chinese ICUs: a national cross-sectional survey. Crit Care Med. 2020;48:e209–18.
Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. The Lancet. 2018;392:75–87.
Gabarin RS, Li M, Zimmel PA, Marshall JC, Li Y, Zhang H. Intracellular and extracellular lipopolysaccharide signaling in sepsis: avenues for novel therapeutic strategies. J Innate Immun. 2021;13:323–32.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7.
Yang D, Zheng X, Wang N, Fan S, Yang Y, Lu Y, et al. Kukoamine B promotes TLR4-independent lipopolysaccharide uptake in murine hepatocytes. Oncotarget. 2016;7:57498–513.
Qin WT, Wang X, Shen WC, Sun BW. A novel role of kukoamine B: Inhibition of the inflammatory response in the livers of lipopolysaccharide-induced septic mice via its unique property of combining with lipopolysaccharide. Exp Ther Med. 2015;9:725–32.
Liu X, Zheng X, Wang N, Cao H, Lu Y, Long Y, et al. Kukoamine B, a novel dual inhibitor of LPS and CpG DNA, is a potential candidate for sepsis treatment. Br J Pharmacol. 2011;162:1274–90.
Zhao Q, Li L, Zhu Y, Hou D, Li Y, Guo X, et al. Kukoamine B ameliorate insulin resistance, oxidative stress, inflammation and other metabolic abnormalities in high-fat/high-fructose-fed rats. Diabetes Metab Syndr Obes. 2020;13:1843–53.
Wang H, Hu X, Wang T, Cui C, Jiang J, Dong K, et al. Exposure-response modeling to support dosing selection for phase IIb development of kukoamine B in sepsis patients. Front Pharmacol. 2021;12: 645130.
Wang H, Wang T, Hu X, Deng C, Jiang J, Qin H, et al. Fixed dosing of kukoamine B in sepsis patients: results from population pharmacokinetic modelling and simulation. Br J Clin Pharmacol. 2022;88:4111–20.
Acknowledgements
The authors would like to thank staff on the study team and all volunteers.
Medical Writing/Editorial Assistance
We did not receive any medical writing or editorial assistance during the writing of this article.
Funding
This study was sponsored by Tian** Chasesun Pharmaceutical Co., Ltd, China. The work was also supported by National High Level Hospital Clinical Research Funding (2022-PUMCH-B-118), National High Level Hospital Clinical Research Funding (No. 2022-PUMCH-A-144), the **qiao Project of Bei**g Association of Science and Technology (No. ZZ19005), National High Level Hospital Clinical Research Funding (No. 2022-PUMCH-B-033), Drug Development and Application of Chinese Pharmacological Society (No. 2019DL001). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. The journal’s Rapid Service Fee was funded by Tian** Chase Sun Pharmaceutical Co.
Author information
Authors and Affiliations
Contributions
Pei Hu, Hongzhong Liu, Qian Zhao, Ji Jiang, Shuai Chen, Kai Kong wrote manuscript, designed protocol, performed data analysis and interpreted results. Hongzhong Liu, Yu** Yuan, Wei Tian, and Chunyan ** were responsible for subject dosing and clinical trial operations. Zhenlei Wang and Teng Wang performed sample testing. Wen Zhong performed statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Hongzhong Liu, Qian Zhao, Yu** Yuan, Zhenlei Wang, Teng Wang, Wei Tian, Wen Zhong, Ji Jiang, and Pei Hu declare no conflicts of interest. Shuai Chen, Kai Kong, and Chunyan ** are full-time employees of Tian** Chasesun Pharmaceutical Co., Ltd.
Ethical Approval
The study was performed in accordance with the International Conference on Harmonization Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki of 1964 and its later amendments. This study was approved by the Ethics Review Board of Peking Union Medical College Hospital (approved no. 2014L01029). All participants provided written informed consent before enrollment.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Liu, H., Zhao, Q., Yuan, Y. et al. First-in-Human Safety, Tolerability, and Pharmacokinetics of Single-Dose Kukoamine B Mesylate in Healthy Subjects: A Randomized, Double-Blind, Placebo-Controlled Phase I Study. Infect Dis Ther 13, 361–371 (2024). https://doi.org/10.1007/s40121-024-00921-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-024-00921-6