
Abstract
The genetic diversity of the honey bee (Apis mellifera) remains undescribed in some parts of its natural distribution range, concretely in those subspecies inhabiting central and southeastern Europe: the globally distributed A. m. ligustica and A. m. carnica, and A. m. cecropia and A. m. macedonica. In this study, we add further information about their genetic variation, differentiation, and admixture using maternal and codominant (microsatellites) molecular markers. The phylogeographic analysis demonstrated the distinct distribution of mitochondrial haplotypes across the sampled geographic range. Cluster analysis detected two main groups of colonies while further discriminant analysis of the principal components supported the existence of ecotypes within A. m carnica and A. m. macedonica, and admixture of them. The extent of introgression and the presence of foreign haplotypes in some of these populations suggested that introductions of non-native subspecies represent a serious threat to the genetic integrity of native honey bee populations due to the creation of hybrid populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The conservation of the honey bee (Apis mellifera L.) subspecies has long been ignored because it is compromised by the activity of beekeepers. Practices such as commercial honey bee breeding, the intensification of queen importation, and transhumance movements change the distribution and diversity of native honey bee populations and ecotypes (De la Rúa et al. 2009), thereby threatening the local populations (De la Rúa et al. 2013). Furthermore, recent dramatic losses in honey bee colonies jeopardize the ecosystem services provided by this key pollinator (Jaffé et al. 2009). Thus, the conservation status of honey bees within their original distribution areas needs to be reconsidered.
The natural distribution range of the honey bee includes Africa, Europe, and Western Asia, but today this species is found worldwide due to multiple migrations and introductions (Moritz et al. 2005; Whitfield et al. 2006). Honey bee populations exhibit differences in morphological (Ruttner 1988), behavioral (Uzunov et al. 2014), and colony development (Dražić et al. 2014; Hat**a et al. 2014) characters due to historical patterns of population isolation and adaptation to the extensive habitat variation found in its natural distribution range. About 30 subspecies of A. mellifera are recognized based on morphometric characters (Ruttner 1988, 1992; Sheppard et al. 1997; Engel 1999; Sheppard and Meixner 2003; Meixner et al. 2011; Chen et al. 2016) of which ten are native to Europe and Mediterranean islands (De la Rúa et al. 2009). These subspecies, which are also described as “geographic races” (Ruttner 1988), have been grouped into four main evolutionary branches by morphometric methods and there is a strong correlation with discrete geographical areas: Near East (O), tropical Africa (A), and Mediterranean/European (M and C) branches (Ruttner 1988). This grou** was confirmed using molecular tools (Cornuet and Garnery 1991; Franck et al. 2000a; Wallberg et al. 2014), increased (Y lineage; Franck et al. 2001; Cridland et al. 2017), and modified in relation to the composition of subspecies (Smith et al. 1997; Sinacori et al. 1998; Kandemir et al. 2006).
Introgression and displacement can occur very fast in honey bee populations, as in contrast to other managed animals, mating is extremely difficult to control in honey bees (Neumann et al. 1999; Palmer and Oldroyd 2000). Therefore gene flow is frequent between neighboring honey bees subspecies and local populations (Franck et al. 1998; Garnery et al. 1998a, b; Sheppard et al. 1991a, b; Jensen et al. 2005). In Europe, apart from the intentional replacement of A. m. mellifera by A. m. carnica in Germany and neighboring countries (Dreher 1946; Maul and Hähnle 1994), the importations and spread of A. m. carnica, A. m. ligustica, and some hybrid strains are mostly known from other regions (Garnery et al. 1998a, b; Ivanova et al. 2007; Jensen et al. 2005; Strange et al. 2008; Bertrand et al. 2015; Henriques et al. 2018). In this sense, the introduction of foreign subspecies into habitats already occupied by native bees exposes the native populations to introgressive hybridization, which can modify the genetic pool of local honey bee populations and drive the loss of their genetic diversity, since particularly rare alleles are lost in a reduced gene pool (De la Rúa et al. 2009; Muñoz et al. 2012a).
Central and southeast European honey bee C-subspecies (A. m. carnica, A. m. ligustica, A. m. macedonica, and A. m. cecropia) are naturally distributed in the Balkan region, the plains of the Danube, and diverse areas in the Apennine Peninsula and Ukraine. Ruttner (1992) found morphometric differences among C-subspecies, but also found local variation within subspecies related to climatic and geographical zones. He described three ecotypes of A. m. carnica morphologically: Alpine (in Austria and northern mountainous Slovenia), Pannonian (in Hungary, Romania to the Carpathians, south Slovenia, Croatia, Serbia, Bosnia and Herzegovina, Montenegro, and Albania), and Dalmatian (in the Adriatic coast), and two ecotypes of A. m. macedonica: Carpathian (in Ukraine and the Romanian plains of the lower Danube River and east of the Carpathian mountains) and Pontic (in Bulgaria, the Former Yugoslav Republic of Macedonia (FYROM) and northern Greece). The natural distribution of A. m. carnica and A. m. macedonica has been shown to be more correlated with climate than with geography, at least in Romania (Coroian et al. 2014).
Areas of hybridization have been detected in C-honey bee populations: in Albania between A. m. carnica and A. m. macedonica (Dedej et al. 1996); in FYROM by the interaction of native A. m. macedonica with A. m. ligustica and A. m. carnica, due to the traditional and long-term intensive importation of A. m. carnica and uncontrolled importation of A. m. ligustica queens (Uzunov et al. 2009); in Bulgaria, where A. m. ligustica, A. m. carnica, and A. m. caucasica have commonly been reared for more than three decades, and strongly hybridized with the native A. m. macedonica (Ivanova et al. 2007); in central Greece, commercial queen breeding and migratory beekee** have contributed to the almost complete hybridization (Bouga et al. 2005) of the four native subspecies originally described by Ruttner (1988). In addition, mitochondrial haplotypes characteristic of A. m. ligustica were detected in the Carniolan honey bee populations from Slovenia (Sušnik et al. 2004), Croatia (Muñoz et al. 2009), and Serbia (see Figure 1 in Nedić et al. 2014 for an overview).

Natural distribution area of A. mellifera subspecies and ecotypes belonging to the east European evolutionary lineage C (adapted and modified from De la Rua et al. 2009). Number of colonies and details by country are shown in Supplementary Information.
To develop measures for the conservation of C-subspecies and ecotypes, it is necessary to characterize the genetic diversity status of at-risk natural populations. Thus, the first goal of the present study was to describe the current genetic diversity of central and southeast European honey bee populations by analyzing mitochondrial and microsatellite markers. Second, we quantified the amount of introgression and hybridization among the populations. Finally, we emphasize the usefulness of molecular tools for strategic conservation practices and new sustainable breeding designs for commercial beekee**.
2 Material and methods
2.1 Honey bee samples
Honey bee samples were collected throughout the C-subspecies distribution in central and southeastern Europe during 2007–2012 (Figures 1 and S5). Honey bee samples were grouped into four datasets related to their geographical distribution: ligustica (honey bee samples from Italy), carnica (samples from Austria, Slovenia, Hungary, Croatia, Bosnia, Montenegro, Albania, Serbia, and Romania), macedonica (samples from southeast Serbia, southeast Romania, FYROM, Bulgaria, and north Greece), and cecropia (samples from the Greek islands and Peloponnese). Adult honey bee workers were sampled from 564 colonies and preserved in absolute ethanol at − 20 °C. Total DNA was extracted from a pair of legs using the Chelex® method (Evans et al. 2013).
2.2 Mitochondrial DNA analysis
The method used to identify the maternal origin of the honey bee colonies and to allocate them to different evolutionary lineages was based on the variation in the mitochondrial intergenic region located between the tRNAleu and cox2 genes (Garnery et al. 1993). Mitochondrial markers can be used to investigate the ancestry of individual colonies (which is inherited directly from the queen). Two types of sequences are present in this region: P and Q. The intergenic region of honey bees from the C-lineage contains a single copy of the Q sequence (Garnery et al. 1993). The combined length and sequence variation detected in each worker honey bee with a restriction fragment length polymorphism or RFLP approach, determined different C-haplotypes (C1 in A. m. ligustica, C2 and C3 in other C-subspecies). Further sequencing of the intergenic region allowed more C-haplotypes (or variants) to be discriminated (named with a lower-case letter following Rortais et al. 2011).
One worker bee per colony was used for mitochondrial DNA (mtDNA) identification. The intergenic tRNAleu-cox2 region was PCR-amplified using a thermocycler PTC 100 (MJ Research) in a total volume of 25 μL with KapaTaq DNA Polymerase (KAPA BIOSYSTEMS), which contained 4 μL of DNA template, 200 μM total dNTP, 1× Reaction Buffer, 0.5 U/rxn KapaTaq DNA Polymerase, 1.5 mM MgCl2, and 0.4 μM of each primer (E2: 5′-GGCAGAATAAGTGCATTG-3′ and H2: 5′-CAATATCATT GATGACC-3′, Garnery et al. 1993). The thermocycler program was as follows: 94 °C (5 min); 35 cycles of a 45-s denaturation at 94 °C, a 45-s elongation at 48 °C, a 60-s extension at 62 °C; and a final extension step at 65 °C for 20 min. Amplicons of each sample were purified with isopropanol and 5 M ammonium acetate and submitted for sequencing (Secugen S.L., Madrid, Spain) using an ABI® PRISM 310 sequencer (Applied Biosystems, Foster City, CA, USA) with E2 and H2 primers.
2.3 Microsatellite genoty**
Biparentally inherited nuclear markers provide powerful information about population events such as introgressive hybridization via mating between drones and queens. A total of 12 polymorphic microsatellite loci (plex 1: A7, A113, Ap43, Ap55, and B124 and plex 2: A8, A79, A88, Ac11, Ap224, Ap249, and Ap274; Estoup et al. 1995; Garnery et al. 1998b; Solignac et al. 2003) were screened using two different multiplex PCRs. The PCR reactions were conducted in a 10 μL total volume that contained 1× reaction buffer (Netline), 1.2 mM MgCl2, 0.3 mM of each dNTP, 0.4 μM of each primer, 3 U Taq polymerase (Netline), and 2 μL of DNA extract. The amplification was performed at 95 °C for one 5-min cycle, 30 cycles of 95 °C for 30 s, 54 °C (plex 1) or 50 °C (plex 2) for 30 s, 72 °C for 30 s, and 72 °C for a 30 min cycle. PCR products of each multiplex were visualized separately by capillary electrophoresis using an ABI® 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA) and sized with an internal size-standard (Servei Central de Suport a la Investigació Experimental, University of Valencia, Spain). Alleles were subsequently scored using GeneMapper® v3.7 (Applied Biosystems, Foster City, CA, USA).
2.4 Genetic diversity indexes
Multiple mitochondrial alignments were produced using the online version of the multiple alignment program for amino acid or nucleotide sequences (MAFFT v7, Katoh and Toh 2008). Sequences of each new and previously published haplotypes were deposited in GenBank. Individual rarefaction curves based on haplotype richness and Shannon H index were generated with the PAST 4.02 program (Hammer et al. 2001) to estimate both sampling sufficiency and expected occurrence of haplotypes for smaller samples. The average number of haplotypes (Nh), number of effective haplotypes (Ne), number of private haplotypes (Np), haplotype diversity (D) and haplotype unbiased diversity (uD) were calculated for all subspecies using GENALEX v6.5 (Peakall and Smouse 2006).
Population genetic parameters based on microsatellite variation were also estimated with GENALEX v6.5 (Peakall and Smouse 2006) using a single honey bee worker genotype per colony. Genetic diversity within regions and subspecies was evaluated by computing the allele frequencies, observed (Ho) and unbiased expected heterozygosity (uHe), and allelic richness (Ar) using rarefaction with the HP-RARE software (Kalinowski 2005). The Hardy-Weinberg equilibrium was tested with Genepop (Raymond and Rousset 1995).
2.5 Population structure and hybridization analyses
A haplotype network was constructed using the median-joining algorithm (Bandelt et al. 1999) with NETWORK v4.6.1.1 (fluxus-engineering.com). A Bayesian model-based clustering method using four sets of samples was performed in an initial attempt to determine whether the sampling regions represented genetically distinct subspecies, which estimated the posterior probability for a given number of genetic populations that resembled subspecies (K) with STRUCTURE v2.3.2 (Pritchard et al. 2000). The set of samples was divided into several data sets so that in three analyses one honey bee per colony was used and in a last case from 1 to 5 honey bees per colony were used, running each one separately in STRUCTURE. An admixture model was used that assumed correlated allele frequencies. The results were based on simulations with 100,000 burn-in steps and 1,000,000 MCMC (Markov Chain Monte Carlo algorithm) iterations. Five runs were used for each K value (K = 1–10) to estimate the most likely value of K. The number of clusters was defined using the value of ΔK described in Evanno et al. (2005) using STRUCTURE HARVESTER (Earl and vonHoldt 2012) and the results of the runs were combined with the software CLUMPP (Jakobsson and Rosenberg 2007). We also used a discriminant analysis of principal components (DAPCs), a multivariate method that allows probabilistic assignment of individuals to each cluster. Unlike STRUCTURE, DAPCs do not require an a priori population genetic model to identify clusters and it is especially suitable for describing clusters of genetically similar individuals (Jombart et al. 2010). DAPC analysis was performed using the “adegenet” package (Jombart 2008) in the statistical program R v2.13.1 (R Development Core Team 2011). The optimal number of DAPC clusters in the data was determined using the diffNgroup option, which identifies sharp changes in the fit of models (measured using the Bayesian Information Criterion) with different numbers of clusters. The individual assignment test was performed using GENALEX to find the proportion of introgressed (hybrid) individuals among C-subspecies. This program uses log-likelihood values to calculate assignments. Also, individual assignment tests were carried out in the program GENECLASS v.2.0 (Piry et al. 2004) using Nei’s standard distance (Nei 1972), a Bayesian method (Rannala and Mountain 1997) and a Bayesian MCMC method (Cornuet et al. 1999) as the criteria for computation. For these analyses, the purest individuals detected in STRUCTURE (average membership proportions Qi > 95%) were selected as reference groups and the assignment of the remaining samples was evaluated.
Genetic differentiation was assessed using FST analysis, which generally performs well when the divergence among samples is expected to be low (Balloux and Goudet 2002). The total genetic differentiation between clusters was assessed using analysis of molecular variance (AMOVA), in addition to pairwise FST estimates (using Weir and Cockerham estimator) implemented in ARLEQUIN v.3.1 (Excoffier et al. 2005). The significance of these estimators was assessed using a non-parametric permutation approach (10,000 permutations) and a Bonferroni correction was applied to significance values.
3 Results
3.1 Genetic diversity
The diversity indices for mtDNA and microsatellites demonstrated high levels of genetic variation within honey bee C-subspecies (Tables I and II respectively). The tRNAleu-cox2 intergenic region from 564 honey bee individuals was characterized by the presence of only one Q sequence, which corresponded to the predicted composition of this region in the C-evolutionary lineage subspecies. Using the RFLP approach, we were able to differentiate three different restriction patterns (with DraI fragment sizes 47-41-64-420: C1, 47-40-64-420: C2, and 47-40-63-420: C3), while the sequence alignment of 578 bp detected 28 polymorphic sites (4.84%) that defined 29 haplotype variants (Table S1). Eighteen of the 29 haplotypes detected were known from previous studies (Franck et al. 2000a; Sušnik et al. 2004; Özdil et al. 2009; Muñoz et al. 2009, 2012b; Coroian et al. 2014) whereas 11 were novel. The mean haplotype unbiased diversity (uD) varied from 0.000 (ligustica set) to 0.758 (carnica set). The individual rarefaction curves (Figure S1) of the haplotype diversity indicated that sampling was representative for the ligustica, carnica, and macedonica sets but not for the cecropia set so this set was used only for reference purposes.
A total of 558 honey bee individuals provided reliable genotypes with the 12 microsatellite loci analyzed. As a first insight into the genetic diversity of central and southeast European honey bees, we inferred population parameters by grou** the samples into sets based on the geographic location of the sampled colonies. All microsatellite loci were highly polymorphic with a total of 186 observed alleles. The overall number of alleles ranged from seven (Ap274) to 40 (A7) and the average number of alleles varied from 4.83 (cecropia set) to 13.83 (carnica set). After rarefaction, the mean allelic and private allelic richness were lower in the ligustica set and higher in the cecropia set. The average genetic diversity, measured as expected heterozygosity (uHe), ranged from 0.612 (carnica) in the north to 0.734 (cecropia) in the south of the Balkan Peninsula, which again was lower in the ligustica set (0.519). Carnica and macedonica sets showed significant deviations from the Hardy-Weinberg equilibrium. These departures were mostly positive, as quantified by FIS values, which indicated an excess of homozygotes at these sites. Significant FIS were observed in every C-subspecies set ranging from 0.06 in carnica to 0.19 in cecropia, although the ligustica set had positive values of FIS.
3.2 Phylogeographic pattern
The most common haplotype was C2d (38.8%), followed by C2c (23.8%) and C1a (11.2%). Differences in the haplotype distribution were observed at subspecies and geographical levels (Table S2). C2d was very frequent in the southeast Balkan Peninsula, while C2c was abundant in the north of the study region. C1a was most common in the Italian Peninsula (ligustica set).
The haplotype network based on mtDNA sequences from 564 samples from four C-subspecies detected a highly complex distribution of haplotypes (Figure 2 and Table S2). The C2d haplotype occupied a central position and haplotypes such as C2b, C2e, C2i, C2j, C2q, C2u, C2v, C2w, C2aa, C2ab, C2ad, and C2ag were connected to C2d by a single mutational change whereas other haplotypes were connected by three or more mutational steps. These haplotypes were distributed in the center and south of the Balkan Peninsula. On the left-hand site, C2c was connected with C2d by C2v (detected in only one colony from Romania), while the haplotypes C1a, C2ac, C2s, C2y, and C2z were connected with C2c by single mutational steps with a star-like topology. C2c and the connected haplotypes were distributed in the north of the Balkan Peninsula. The most common Italian haplotype C1a was connected with C3a (detected in only two colonies from Slovenia) and C2c (distributed in the Italian Peninsula and some northern countries in the Balkan Peninsula) by one mutational change.

Median-joining network based on 578-bp sequences of the tRNAleu-cox2 intergenic region for 564 honey bee colonies from eastern Europe. The size of the circles is proportional to the number of sequences assigned to that haplotype. Small black line on link connecting the haplotype indicates one mutational difference. A small black square represents median vectors.
3.3 Population genetic structure
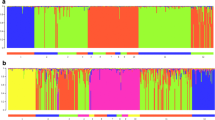
The STRUCTURE simulations with the four samples sets (Figure S2) showed that the optimal number of clusters was two after correction based on Evanno et al. (2005) (for all analyzed honey bee workers: K = 2, mean LnP(D) = − 31,455.42, mean Var[LnP(D)] = 320.15, Figure S2D). At K = 2, honey bee populations from the Italian Peninsula (A. m. ligustica) and the northern and central honey bee populations from the Balkan Peninsula (A. m. carnica) were assigned to cluster 1, while southern honey bee populations from the Balkan Peninsula (A. m. macedonica) and some from Greek islands (A. m. cecropia) were included in cluster 2. Clear signs of admixture were observed between these clusters, mostly in Romania and Serbia. However, from a biogeographical perspective, the optimal model was rather six genetic clusters in which a genetic substructure appeared (Figure 3); therefore, we also analyzed K = 3–6 with STRUCTURE. This genetic substructure was confirmed by independent runs with the four sample sets (Figure S3). At K = 3, we observed three groups, cluster 1 formed mainly by the samples from Italy corresponding to A. m. ligustica and the northwest of Balkan Peninsula (Austria, Slovenia, Hungary, and Croatia), cluster 3 formed by the samples from southern of Balkan Peninsula (Bulgaria and Greece), and the cluster 2 with admixture formed mainly by honey bees from the central of the Balkan Peninsula. At K = 4, we found additional substructure within honey bee populations from the central of the Balkan Peninsula, with a fourth cluster formed by the samples from southeast Serbia and FYROM. At K = 5, a new cluster formed mainly by the samples of bees from Greece can be observed, and at K = 6 appears another more heterogeneous one within the samples from the northwest of the Balkan Peninsula.

Results of STRUCTURE analysis based on microsatellite data of all honey bee workers (1–5 individuals per colony) assuming from K = 2 to K = 6. Each individual is represented by a single vertical line, divided into K colors. The colored segment shows the individual’s estimated proportion of membership to the genetic cluster. IT, Italy; AT, Austria; SI, Slovenia; HU, Hungary; HR, Croatia; BA, Bosnia and Herzegovina; ME, Montenegro; RO, Romania; RS, Serbia; RO-SE, southeast of Romania; RS-SE, southeast of Serbia; AL, Albania; MK, Macedonia; BU, Bulgaria; EL, Greece.
These six STRUCTURE clusters were further corroborated by DAPCs (Figure 4). In the analyses of K = 4, the samples from Bulgaria and Greece (cluster iv) appeared to be separated from the other three clusters. The separate examination with DAPCs using the two main clusters inferred by STRUCTURE detected three clusters (Figure 4b) in STRUCTURE-based cluster 1 (honey bee samples from the Italian Peninsula and the center north Balkan Peninsula) which corresponded to ligustica (cluster 1: Italian Peninsula) and two clusters within carnica subspecies, the carnica-1 (cluster 2) with honey bees from northwest of Balkan Peninsula (Austria, Slovenia, Hungary, Croatia, Bosnia and Herzegovina, Serbia-north and Romania-north) and the carnica-2 (cluster 3) mainly with honey bees from central and east of Balkan Peninsula (Montenegro, Serbia, and Romania). As well as three different clusters (Figure 4c) in STRUCTURE-based cluster 2 (southern honey bee populations from Balkan Peninsula), which corresponded to two clusters within the macedonica subspecies (honey bee samples from the southeast of Serbia and FYROM in cluster 5/macedonica-1, and samples from Bulgaria and the north of Greece in cluster 6/macedonica-2), and finally one cluster of cecropia subspecies from the south of Greece and Greek islands (cluster 4).

Inference of genetic clusters using discriminant analysis of PCs (DAPCs) based on microsatellite data for four clusters with all honey bee samples (a), the DAPC results for the STRUCTURE-based cluster 1 formed only with the honey bee samples from ligustica and carnica of the clusters i and ii of panel a (b), and the DAPC results for the analysis for the STRUCTURE-based cluster 2 with only the samples from clusters iii and iv of panel a (c). The up-left plot shows the Bayesian Information Criterion (BIC) using the diffNgroup option for different numbers of clusters (K), with the optimal K for panel a indicated by a red circle.
3.4 Introgression and hybridization
The STRUCTURE analysis indicated a high proportion of admixture in the honey bee populations from Romania and Serbia. To obtain further insights into the level of hybridization in these European honey bee populations, we conducted an assignment test in GENALEX using the clusters defined by DAPCs (Table III). The clusters with the highest percentage of individuals assigned to their own population were macedonica-1 (97.18%), ligustica (93.59%), and macedonica-2 (89.41%). The populations of carnica had a higher level of ligustica introgression (6.11 and 2.16% in carnica-1 and carnica-2 respectively), while the population of cecropia displayed introgression from carnica (4.65%) and macedonica-2 (32.56%). The percentages of introgressed individuals between carnica groups were greater than those detected between macedonica groups.
In the assignment test GENECLASS using the purest individuals detected in STRUCTURE (average membership proportions Qi > 95%) as reference groups, we were able to correctly assign 92.91% and 55.35% of all simulated individuals to their respective reference populations using the Nei’s standard distance and Rannala and Mountain methods respectively (Figure 5). Comparing both methods, the Nei’s standard distance (Nei 1972) allowed to assign more individuals than the Bayesian method (Rannala and Mountain 1997). For both methodologies, the population of Italy was assigned to the ligustica group, those of southeast of Serbia and FYROM to the macedonica-1 group, and those of Bulgaria and northern Greece to the macedonica-2 group. However, the assignment of the Austrian individuals was more ambiguous, finding the same probability of belonging to carnica-1 and carnica-2 groups by both methods. For the remaining northern and central Balkan populations, the highest probability value for the Nei method corresponded to the population assignment of the Bayesian method, assigning the Hungarian population to the carnica-2 group and the remaining populations to the carnica-1 group.

Distribution of highest probability assignments of honey bee individuals from each geographical origin to belong to the defined subspecies and ecotypes as determined using the resampling procedure in GENECLASS 2.0 (Piry et al. 2004) by Nei’s standard distance (Nei 1972) method (bars), and by Bayesian (Rannala and Mountain 1997) method (points).
The histogram of the hybrid individuals detected for each inferred cluster using DAPCs (Figure S4a) revealed a higher amount of hybrid individuals in the carnica clusters (26.52 and 27.08% in carnica-1 and carnica-2 respectively). The inferred genetic composition of the hybrid individuals (Figure S4b) showed that the majority had a higher proportion of ligustica alleles while the carnica groups had a considerable hybridization among them. The graphical representation of the subspecies and clusters distribution in the sampled region shows this complex pattern (Figure S5).
3.5 Genetic diversity within DAPC clusters
The allelic pattern based on the 12 microsatellite loci in the six clusters detected using DAPCs is shown in Figure 6. The cluster carnica-1 had the highest number of private alleles. The number of alleles decreased from north to south while the mean values of genetic diversity (uHe) showed the opposite trend, decreasing from the south of Greece (A. m. macedonica) to the north of the Balkan peninsula (carnica clusters) while the lowest values were found in the Italian peninsula. As in the previous analysis at the subspecies (set) level, the carnica and macedonica clusters had significant deviations from the Hardy-Weinberg equilibrium.

Allelic patterns for microsatellite data in C-subspecies and detected clusters within A. m. carnica and A. m. macedonica subspecies with DAPC.
The clusters obtained from the DAPC analysis exhibited significant genetic differentiation according to the pairwise FST comparison (Table IV). The FST value between carnica clusters (0.054) was lower than that between macedonica clusters (0.121), while the ligustica and cecropia clusters had minor significant FST values with carnica-1 (0.075) and macedonica-2 (0.066), respectively. The proportions of genetic variation associated with the carnica and macedonica DAPCs clusters were significantly different (Table V) and there was a significant substructure within carnica (5.28%) and macedonica (8.81%) subspecies based on the microsatellite data.
4 Discussion
Our results revealed extensive intraspecific hybridization among the four different subspecies of honey bees belonging to the central and southeast Europe evolutionary C-lineage. In some populations, the mtDNA signal of introduced subspecies was present in one-third of the colonies sampled. However, signs of natural genetic variation could still be traced by analyzing codominant markers. Overall, our results confirmed the hypothesis of the existence of local ecotypes within this lineage, so it is reasonable to state that the introductions of non-native honey bee queens into these regions represent a serious threat to the genetic integrity of local populations due to introgressive hybridization.
4.1 Genetic diversity and differentiation within subspecies
Compared with other European honey bee populations that correspond to other (Jensen et al. 2005; Cánovas et al. 2011; Rortais et al. 2011; Pinto et al. 2014) or the same (Soland-Reckeweg et al. 2008; Dall’Olio et al. 2007) evolutionary lineages, the central and southeastern honey bee populations had a high level of genetic diversity at the mitochondrial and nuclear (microsatellite) levels. The mtDNA data showed that the haplotype genetic diversity was higher in the Balkans than in the Italian Peninsula (Franck et al. 2000b). In contrast to the West European honey bee, A. m. mellifera (Jensen et al. 2005; Pinto et al. 2014), a phylogeographic pattern irrespective of introgression based on the presence of foreign haplotypes in these populations can still be observed. These contrasting results may have been due to the lack of geographic barriers to gene flow in the former region and the presence of mountain ranges running from north-west to south-east in the Balkan Peninsula. Despite this natural pattern, the close connections among beekeepers were also reflected in our results because the queen trade is fairly intensive in this area and introductions of queens with different origins could be the source of the extreme haplotype variation detected.
Microsatellite markers were more effective for detecting genetic diversity changes and subsequent population structuring. Our results demonstrated that the southern honey bee populations are more diverse than the northern ones. This agrees with studies of Spanish and French populations of the West European honey bee (Garnery et al. 1998b) where A. m. mellifera populations exhibited a diversity cline from south to north, which suggested that this subspecies has naturally colonized northern Europe from its southern refuge in Spain (Chávez-Galarza et al. 2015). The higher level of genetic diversity detected in this study also suggests that the colonization of east Europe involved honey bees from the Balkan Peninsula. This colonization pattern has been observed in other species, where northern Europe has been colonized at least partially from refugia in the Balkans or areas further east (Hewitt 1999, 2000).
In the eastern C-lineage, the substructure was partially in agreement with the original subspecies and ecotype definitions based on the morphological and molecular descriptions of Ruttner (1988, 1992) and Muñoz et al. (2009). For A. m. carnica, Ruttner (1992) described the presence of two morphologically distinct ecotypes according to zoogeographical zones, i.e., the Pannonian and the Alpine ecotypes, and one variation spread along the Adriatic coast, the Dalmatian. Our results confirmed the existence of two groups in A. m. carnica, i.e., carnica-1 and carnica-2, which may be associated with the Pannonian and Alpine ecotypes, respectively. Although significant, the amount of differentiation between them is small and considerable gene flow and hybridization between the carnica populations was detected that led to partial homogenization of the two carnica ecotypes. For A. m. macedonica, Ruttner (1988, 1992) described two morphological ecotypes: the Carpathian in Ukraine and Romania in the plains of the lower Danube River and east of the Carpathian Mountains, and the Pontic ecotype in Bulgaria, FYROM, and northern Greece. Our results demonstrated the presence of two clusters within A. m. macedonica, i.e., macedonica-1 and macedonica-2, in the distribution area of the Pontic ecotype. The cluster macedonica-1 was found in the southeast of Serbia and FYROM, whereas the macedonica-2 group was detected in Bulgaria and northern Greece. In this case, the genetic divergence, high percentage of individuals re-assigned to self-cluster, and the low number of hybrid individuals suggested restricted gene flow between honey bee populations belonging to the macedonica ecotypes. These results are consistent with those obtained by Nedić et al. (2014) who found that in Serbia, there was a clinal segregation between A. m. carnica in the northwest and A. m. macedonica in the southeast, and also by Uzunov et al. (2014) who found that the honey bee population from Bulgaria was different from the other samples analyzed within the A. m. macedonica subspecies.
4.2 Introgression and implications for conservation
The mtDNA and microsatellite analyses provided evidence for introgression events from neighboring A. mellifera subspecies. Mitochondrial introgression from Italian A. m. ligustica (haplotype C1a) was detected in the northern Balkan Peninsula and the results of the assignment test also indicated nuclear introgression, particularly in the carnica ecotypes. Italian honey bee breeders widely export A. m. ligustica queens inside and outside Italy, and most come from Emilia-Romagna where the C1a haplotype is very common (Franck et al. 2000b; Pinto et al. 2014). This explains the presence of the alien C1a haplotype in many populations from other subspecies and lineages (De la Rúa et al. 1998, 2006; Ferreira et al. 2020; Garnery et al. 1998b; Muñoz and De la Rúa 2012; Muñoz et al. 2009, 2013). Introgression was also higher in the honey bee populations from southern Greece and Greek islands, where 32.56% of individuals were assigned to the macedonica-2 group. This introgression could be a consequence of natural migration or human activity. In addition, we found support for hybridization, specifically between carnica ecotypes and between ligustica and carnica ecotypes.
Hybridization is not reversible and will produce a complete admixture of ecotypes or subspecies (Allendorf and Luikart 2007), but high proportions of pure A. mellifera C-subspecies were still found. In the case of A. m. ligustica, the occurrence of pure individuals could be due to the use of ligustica queens from breeding programs to export Italian queens around the world (Dall’Olio et al. 2007; Fontana et al. 2018). High levels of pure individuals were also observed in two differentiated ecotypes of A. m. macedonica in southeastern Europe. A. m. carnica is significantly genetic different from neighboring subspecies, but a considerable number of hybrid individuals were found. The formation of hybrid populations could produce the observed admixture. However, a significant genetic divergence between the two detected ecotypes could still be traced despite the intense hybridization process between them. In the case of A. m. cecropia, subspecies introgression from A. m. carnica and A. m. macedonica has occurred to varying degrees among subspecies, although this observation should be taken with caution given the small sample size of individuals of this subspecies analyzed.
To increase the effectiveness of conservation areas and pure breeding populations or ecotypes, more information is required about the honey bee genetic biodiversity of central and southeast of Europe to avoid the genetic homogenization of honey bee ecotypes. Topographic structures such as mountains and large water bodies have been proposed to inhibit the flight paths of drones (De la Rúa et al. 2002) so they could prevent the mating of native queens with drones from foreign introduced colonies. Moreover, the genetic identification of hybrid individuals could be a useful tool for excluding introgressed and hybrid individuals from conservation breeding programs (Soland-Reckeweg et al. 2008; Muñoz and De la Rúa 2012; Henriques et al. 2019).
We have confirmed that local ecotypes are still extant in central and southeast European honey bee populations. The effect of intraspecific hybridization may drive the loss of these ecotypes (Allendorf et al. 2001), which constitutes a serious threat to the genetic integrity and persistence of populations that are locally adapted to their environment conditions (Rhymer and Simberloff 1996). Therefore, conservation activities should primarily focus on the prevention of further human-mediated introductions of non-native honey bee queens.
Change history
09 April 2021
A Correction to this paper has been published: https://doi.org/10.1007/s13592-021-00856-8
References
Allendorf, F. W., Luikart, G. (2007) Conservation and the genetics of population, Blackwell Publishing, Malden, MA
Allendorf, F. W., Leary, R. F., Spruell, P., Wenburg, J. K. (2001) The problems with hybrids: setting conservation guidelines. Trends Ecol. Evol. 16 (11), 613-622
Bandelt, H. J., Forster, P., Rohl, A. (1999) Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37-8
Balloux, F., Goudet, F. (2002) Statistical properties of population differentiation estimators under stepwise mutation in a finite island model. Mol. Ecol. 11, 771-783
Bertrand, B., Alburaki, M., Legout, H., Moulin, S., Mougel, F., Garnery, L. (2015). Mt DNA COI‐COII marker and drone congregation area: An efficient method to establish and monitor honeybee (Apis mellifera L.) conservation centres. Mol. Ecol. Res. 15, 673-683
Bouga, M., Harizanis, P. C., Kilias, G., Alahiotis, S. (2005) Genetic Divergence and Phylogenetic Relationships of Honey Bee Apis mellifera (Hymenoptera: Apidae) Populations from Greece and Cyprus using PCR - RFLP Analysis of Three mtDNA Segments. Apidologie 36, 335-344
Cánovas, F., De la Rúa, P., Serrano, J., Galián, J. (2011) Microsatellite variability reveals beekee** influences on Iberian honeybee populations. Apidologie 42, 235-251
Chávez-Galarza, J., Henriques, D., Johnston, J. S., Carneiro, M., Rufino, J., Patton, J. C., Pinto, M. A. (2015). Revisiting the Iberian honey bee (Apis mellifera iberiensis) contact zone: maternal and genome-wide nuclear variations provide support for secondary contact from historical refugia. Mol. Ecol. 24, 2973-2992.
Chen, C., Liu, Z., Pan, Q., Chen, X., Wang, H., Guo, H., Shi, W. (2016) Genomic analyses reveal demographic history and temperate adaptation of the newly discovered honey bee subspecies Apis mellifera sinisxinyuan n ssp. Mol. Biol. Evol. 33, 1337-1348
Cornuet, J. M., Garnery, L. (1991) Mitochondrial-DNA variability in honeybees and its phylogeographic implications. Apidologie 22, 627-642
Cornuet, J.M., Piry, S., Luikart, G., Estoup, A., Solignac, M. (1999) New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153, 1989-2000
Coroian, C. O., Muñoz, I., Schlüns, E. A., Paniti-Teleky, O. R., Erler, S., Furdui, E. M., ... Moritz, R. F. A. (2014). Climate rather than geography separates two European honeybee subspecies. Mol. Ecol. 23, 2353-2361.
Cridland, J. M., Tsutsui, N. D., Ramírez, S. R. (2017). The complex demographic history and evolutionary origin of the western honey bee, Apis mellifera. Genome Biol. Evol. 9, 457-472.
Dall’Olio, R., Marino, A., Lodesani, M., Moritz, R. F. A. (2007). Genetic characterization of Italian honeybees, Apis mellifera ligustica, based on microsatellite DNA polymorphisms. Apidologie 38, 207-217
De la Rúa, P., Serrano, J., Galián, J. (1998) Mitochondrial DNA variability in the Canary Island honeybees (Apis mellifera L). Mol. Ecol. 7, 1543-1548
De la Rúa, P., Galián, J., Serrano, J. (2002) Biodiversity of Apis mellifera populations from Tenerife (Canary Islands) and hybridisation with East European races. Biodiver. Conserv. 11, 59-67
De la Rúa, P., Galián, J., Pedersen, B. V., Serrano, J. (2006) Molecular characterization and population structure of Apis mellifera from Madeira and the Azores. Apidologie 37, 699-708
De la Rúa, P., Jaffé, R., Dall’Olio, R., Muñoz, I., Serrano, J. (2009) Biodiversity, conservation and current threats to European honeybees. Apidologie 40, 263-284
De la Rúa, P., Jaffé, R., Muñoz, I., Serrano, J., Moritz, R. F.A., Kraus, F. (2013) Conserving genetic diversity in the honey bee Comments on Harpur et al (2012). Mol. Ecol. 22, 3208-3210
Dedej, S., Biasiolo, A., Piva, R. (1996) Morphometric and alloenzyme characterization in the Albanian honeybee population Apis mellifera L. Apidologie 27, 121-131
Dražić, M. M., Filipi, J., Prđun, S., Bubalo, D., Špehar, M., Cvitković, D., ... Kezić, N. (2014). Colony development of two Carniolan genotypes (Apis mellifera carnica) in relation to environment. J. Apic. Res. 53, 261-268.
Dreher, K. (1946) Gedanken zum Neuaufbau des Zuchtwesens. Die Hessische Biene 81, 62-64
Earl, D. A., vonHoldt, B. M. (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4 (2), 359-361
Engel, M. S. (1999) The taxonomy of recent and fossil honey bees (Hymenoptera: Apidae, Apis). J. Hymenop. Res. 8, 165-196
Estoup, A., Garnery, L., Solignac, M., Cornuet, J. M. (1995) Microsatellite variation in honey bee (Apis mellifera L) populations: Hierarchical genetic structure and test of the infinite allele and stepwise mutation models. Genetics 140, 679-695
Evanno, G., Regnaut, S., Goudet, J. (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611-2620
Evans, J. D., Schwarz, R. S., Chen, Y. P., Budge, G., Cornman, R. S., De la Rúa, P., ... Genersch, E. (2013). Standard methods for molecular research in Apis mellifera. J. Apic. Res. 52, 1-54.
Excoffier, L., Laval, G., Schneider, S. (2005) Arlequin (version 30): an integrated software package for population genetics data analysis. Evol. Bioinformatic Online 1, 47-50
Ferreira, H., Henriques, D., Neves, C. J., Machado, C. A., Azevedo, J. C., Francoy, T. M., Pinto, M. A. (2020). Historical and contemporaneous human-mediated processes left a strong genetic signature on honey bee populations from the Macaronesian archipelago of the Azores. Apidologie 51, 316-328.
Franck, P., Garnery, L., Solignac, M., Cornuet, J. M. (1998) The origin of west European subspecies of honeybees (Apis mellifera): New insights from microsatellite and mitochondrial data. Evolution 52, 1119-1134
Franck, P., Garnery, L., Solignac, M., Cornuet, J. M. (2000a) Molecular confirmation of a fourth lineage in honeybees from the Near East. Apidologie 31, 167-180
Franck, P., Garnery, L., Celebrano, G., Solignac, M., Cornuet, J. M. (2000b) Hybrid origins of honey bees from Italy (Apis mellifera ligustica) and Sicily (A m sicula). Mol. Ecol. 9, 907-921
Franck, P., Garnery, L., Loiseau, A. (2001) Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity 86, 420-430
Fontana, P., Costa, C., Di Prisco, G., Ruzzier, E., Annoscia, D., et al. (2018). Appeal for biodiversity protection of native honey bee subspecies of Apis mellifera in Italy (San Michele all’Adige declaration). B. Insectol. 71, 257-271,
Garnery, L., Solignac, M., Celebrano, G., Cornuet, J. M. (1993) A simple test using restricted PCR-amplified mitochondrial DNA to study the genetic structure of Apis mellifera L. Experientia 49, 1016-1021
Garnery, L., Franck, P., Baudry, E., Vautrin, D., Cornuet, J. M., Solignac, M. (1998a) Genetic diversity of the west European honey bee (Apis mellifera mellifera and Apis mellifera iberica) I Mitochondrial DNA. Genet. Sel. Evol. 30, 31-47
Garnery, L., Franck, P., Baudry, E., Vautrin, D., Cornuet, J. M., Solignac, M. (1998b) Genetic biodiversity of the west European honeybee (Apis mellifera mellifera and Apis mellifera iberica) II Microsatellite loci. Genet. Sel. Evol. 30, 49-74
Hammer, Ø., Harper, D.A.T., Ryan P. D. (2001) PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electronica 4(1), 9.
Hat**a, F., Costa, C., Büchler, R., Uzunov, A., Drazic, M., Filipi, J., ... Bienkowska, M. (2014). Population dynamics of European honey bee genotypes under different environmental conditions. J. Apic. Res. 53, 233-247
Henriques, D., Browne, K. A., Barnett, M. W., Parejo, M., Kryger, P., et al (2018) High sample throughput genoty** for estimating C-lineage introgression in the dark honeybee: an accurate and cost-effective SNP-based tool. Sci. Rep.-UK 8, 8552
Henriques, D., Parejo, M., Lopes, A. R., Pinto, M. A. (2019). Mitochondrial SNP markers to monitor evolutionary lineage ancestry in Apis mellifera mellifera conservation programs. Apidologie 50, 538-541.
Hewitt, G. M. (1999) Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 68, 87-112
Hewitt, G. M. (2000) The genetic legacy of the quaternary ice ages. Nature 405, 907-913
Ivanova, E. N., Staykova, T. A., Bouga, M. (2007) Allozyme variability in honey bee populations from some mountainous regions in the southwest of Bulgaria. J. Apic. Res. 46, 3-7
Jaffé, R., Dietemann, V., Crewe, R. M., Moritz, R. F. A. (2009) Temporal variation in the genetic structure of a drone congregation area: An insight into the population dynamics of wild African honeybees (Apis mellifera scutellata). Mol. Ecol. 18, 1511-1522
Jakobsson, M. and Rosenberg, N.A. (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801-1906
Jensen, A. B., Palmer, K. A., Boomsma, J. J., Pedersen, B. V. (2005) Varying degrees of Apis mellifera ligustica introgression in protected populations of the black honeybee, Apis mellifera mellifera, in northwest Europe. Mol. Ecol. 14, 93-106
Jombart, T. (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403-1405
Jombart, T., Devillard, S., Balloux, F. (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11, 94
Kalinowski, S. T. (2005) HP-Rare: a computer program for performing rarefaction on measures of allelic diversity. Mol. Ecol. Notes 5, 187-189
Kandemir, I., Meixner, M. D., Ozkan, A., Sheppard, W. S. (2006) Genetic characterization of honey bee (Apis mellifera cypria) populations in northern Cyprus. Apidologie 37, 547-555
Katoh, K., Toh, H. (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform, 9, 286-298
Maul, V., Hähnle, A. (1994) Morphometric studies with pure bred stock of Apis mellifera carnica from Hessen. Apidologie 25, 119-132
Meixner, M. D., Leta, M. A., Koeniger, N., Fuchs, S. (2011) The honey bees of Ethiopia represent a new subspecies of Apis mellifera-Apis mellifera simensis n ssp. Apidologie 42, 425-437
Moritz, R. F. A., Härtel, S., Neumann, P. (2005): Global invasions of the western honey bee (Apis mellifera) and the consequences for biodiversity. Ecoscience 12, 289-301
Muñoz, I., De la Rúa, P. (2012) Temporal analysis of the genetic diversity in a honey bee mating area of an island population (La Palma, Canary Islands, Spain). J. Apic. Sci. 55, 141-148
Muñoz, I., Dall’Olio, R., Lodesani, M., De la Rúa, P. (2009) Population genetic structure of coastal Croatian honeybees (Apis mellifera carnica). Apidologie 40, 617-626
Muñoz, I., Madrid-Jiménez, M. J., De la Rúa, P. (2012a) Temporal genetic analysis of an introgressed island honey bee population (Tenerife, Canary Islands, Spain). J. Apic. Res. 51, 144-146
Muñoz, I., Stevanovic, J., Stanimirovic, Z., De la Rúa, P. (2012b) Genetic variation of Apis mellifera from Serbia inferred from mitochondrial analysis. J. Apic. Sci. 56, 59-69
Muñoz, I., Pinto, M. A., De la Rúa, P. (2013) Temporal changes in mitochondrial diversity highlights contrasting population events in Macaronesian honey bees. Apidologie 44, 295-305
Nedić, N., Francis, R.M., Stanisavljević, L., Pihler, I., Kezić, N., Bendixen, C., Kryger, P. (2014) Detecting population admixture in honey bees of Serbia. J. Apic. Res. 53, 303-313
Nei, M. (1972) Genetic distance between populations. American Naturalist 106: 283-292
Neumann, P., Moritz, R. F. A., Van Praagh, J. P. (1999) Queen mating frequency in different types of honey bee mating apiaries. J. Apic. Res. 38, 11-18
Özdil, F., Yildiz, M. A., Hall, H. G. (2009) Molecular characterization of Turkish honey bee populations (Apis mellifera L) inferred from mitochondrial DNA RFLP and sequence results. Apidologie 40, 570-576
Palmer, K. A., Oldroyd, B. P. (2000). Evolution of multiple mating in the genus Apis. Apidologie 31, 235-248.
Peakall, R., Smouse, P. E. (2006) GENALEX 6: genetic analysis in Excel Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288-295
Pinto, M. A., Henriques, D., Chávez-Galarza, J., Kryger, P., Garnery, L., et al. (2014). Genetic integrity of the Dark European honey bee (Apis mellifera mellifera) from protected populations: a genome-wide assessment using SNPs and mtDNA sequence data. J. Apic. Res. 53, 269-278.
Piry, S., Alapetite, A., Cornuet, J. M., Paetkau, D., Baudouin, L., Estoup, A. (2004) GENECLASS 2: a software for genetic assignment and first-generation migrant detection. J. Hered. 95, 536-539
Pritchard, J. K., Stephens, M., Donnelly, P. J. (2000) Inference of population structure using multilocus genotype data. Genetics 155, 945-959
R Development Core Team (2011) R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna ISBN 3-900051-07-0
Rannala, B., Mountain, J.L. (1997) Detecting immigration by using multilocus genotypes. PNAS 94, 9197-9201
Raymond, M., Rousset, F. (1995) GENEPOP (version 12): population genetics software for exact tests and ecumenicism. J. Hered. 86, 248-249
Rhymer, J. M., Simberloff, D. (1996) Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 27, 83-109
Rortais, A., Arnold, G., Alburaki, M., Legout, H., & Garnery, L. (2011). Review of the DraICOI-COII test for the conservation of the black honeybee (Apis mellifera mellifera). Cons. Gen. Res. 3, 383-391.
Ruttner, F. (1988) Biogeography and Taxonomy of Honeybees, Springer Verlag, Berlin
Ruttner, F. (1992) Naturgeschichte der Honigbienen, Ehrenwirth, Munich
Sheppard, W. S., Meixner, M. D. (2003) Apis mellifera pomonella, a new honey bee subspecies from Central Asia. Apidologie 34, 367-375
Sheppard, W. S., Rinderer, T. E., Mazzoli, J., Stelzer, J. A., Shimanuki, H. (1991a) Gene flow between African- and European-derived honey bee populations in Argentina. Nature 349, 782-784
Sheppard, W. S., Soares, A. E. E., DeJong, D. (1991b) Hybrid status of honey bee populations near the historic origin of Africanization in Brazil. Apidologie 22, 643-52
Sheppard, W. S., Arias, M. C., Grech, A., Meixner, M. D. (1997) Apis mellifera ruttneri, a new honey bee subspecies from Malta. Apidologie 28, 287-293
Sinacori, A., Rinderer, T. E., Lancaster, V., Sheppard, W. S. (1998) A morphological assessment of Apis mellifera from Palermo, Italy. Apidologie 29, 481-490
Smith, D. R., Crespi, B. J., Bookstein, F. L. (1997) Fluctuating asymmetry in the honey bee, Apis mellifera: effects of ploidy and hybridization. J. Evol. Biol. 10, 551-574
Soland-Reckeweg, G., Heckel, G., Neumann, P., Fluri, P., Excoffier, L. (2008) Gene flow in admixed populations and implications for the conservation of the Western honeybee, Apis melliferaJ. Insect Conserv. 13, 317-328
Solignac, M., Vautrin, D., Loiseau, A., Mougel, F., Baudry, E. (2003) Five hundred and fifty microsatellite markers for the study of the honeybee (Apis mellifera L) genome. Mol. Ecol. Notes 3, 307-311
Strange, J. P., Garnery, L., Sheppard, W. S. (2008) Morphological and molecular characterization of the Landes honey bee (Apis mellifera L) ecotype for genetic conservation. J. Insect Conserv. 12, 527-537
Sušnik, S., Kozmus, P., Poklukar, J., Megli, V. (2004) Molecular characterisation of indigenous Apis mellifera carnica in Slovenia. Apidologie 35, 623-636
Uzunov, A., Kiprijanovska, H., Andonov, S., Naumovski, M., Gregorc, A. (2009) Morphological diversity and racial determination of the honey bee (Apis mellifera L) population in the Republic of Macedonia. J. Apic. Res. 48, 196-203
Uzunov, A., Meixner, M. D., Kiprijanovska, H., Andonov, S., Gregorc, A., Ivanova, E., Bouga, M., Dobi, P., Büchler, R., Francis, R., Kryger, P. (2014). Genetic structure of Apis mellifera macedonica in the Balkan Peninsula based on microsatellite DNA polymorphism. J. Apic. Res. 53, 288-295
Wallberg, A., Han, F., Wellhagen, G., Dahle, B., Kawata, M., et al. (2014) A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nature Genetics 46, 1081
Whitfield, C. W., Behura, S. K., Berlocher, S. H., Clark, A. G., Johnston, J. S, et al. (2006) Thrice out of Africa, Ancient and recent expansions of the honey bee, Apis mellifera. Science 314, 642-645
Acknowledgments
We are indebted to institutions and people who kindly provided the honey bee samples: Raffaele Dall’Olio (Italy), Jevrosima Stevanovic (Serbia), Evgeniya Ivanova (Bulgaria), Maria Bouga (Greece), Jutta Vollmann (Austria), Cristian Coroian (Romania), Eliza Cauia (Romania), and Szilvia Kusza (Hungary). The authors wish to thank O. Sánchez and A. I. Asensio for their technical support. Finally, we would like to thank the valuable comments of the referees who have undoubtedly improved this work.
Funding
This research was funded by Fundación Séneca (grants 11961/PI/09 and 19908/GERM/2015) to P. De la Rúa. I. Muñoz was supported by a post-doctoral fellowship “Saavedra Fajardo” (20036/SF/16) and a MINECO Spanish postdoctoral grant “Juan de la Cierva-Incorporación” (JCI2018-036614-I).
Author information
Authors and Affiliations
Contributions
This work formed part of the PhD thesis of IM supervised by PDlR. Both authors designed the experiments. IM extracted DNA and performed genoty** and data analysis. PDlR provided samples. IM wrote the paper with suggestions from PDlR and both read and approved the article prior to publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Manuscript editor: Marina Meixner
L’introgression menace la grande diversité génétique des abeilles mellifères de l’Ancien Monde.
Apis mellifera / Europe centrale et du sud-est / conservation / écotypes / diversité génétique.
Die hohe genetische Diversität bei Altwelt-Honigbienen wird durch Introgression bedroht.
Apis mellifera / Zentral- und Südwest-Europa / Ökotypen / Naturschutz / genetische Diversität.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 2.59 mb)
Rights and permissions
About this article

Cite this article
Muñoz, I., De la Rúa, P. Wide genetic diversity in Old World honey bees threaten by introgression. Apidologie 52, 200–217 (2021). https://doi.org/10.1007/s13592-020-00810-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-020-00810-0