
Abstract
Despite advances in our understanding of the molecular landscape of prostate cancer and the development of novel biomarker-driven therapies, the prognosis of patients with metastatic prostate cancer that is resistant to conventional hormonal therapy remains poor. Data suggest that a significant proportion of patients with metastatic castration-resistant prostate cancer (mCRPC) have mutations in homologous recombination repair (HRR) genes and may benefit from poly(ADP-ribose) polymerase (PARP) inhibitors. However, the adoption of HRR gene mutation testing in prostate cancer remains low, meaning there is a missed opportunity to identify patients who may benefit from targeted therapy with PARP inhibition, with or without novel hormonal agents. Here, we review the current knowledge regarding the clinical significance of HRR gene mutations in prostate cancer and discuss the efficacy of PARP inhibition in patients with mCRPC. This comprehensive overview aims to increase the clinical implementation of HRR gene mutation testing and inform future efforts in personalized treatment of prostate cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Worldwide, prostate cancer accounts for over 7% of all new cancer cases, making it the fourth most common cancer and the second most common cancer in men. |
However, prostate cancer is highly biologically, clinically, and molecularly heterogeneous, leading to an array of treatment options including novel hormonal agents and recent approvals of poly(ADP-ribose) polymerase (PARP) inhibitors, with the latter particularly effective in patients with a deficiency in the homologous recombination repair (HRR) pathway. |
We comprehensively review current literature regarding the role of the HRR pathway in prostate cancer, testing for HRR mutations, and the prognostic and predictive significance of HRR mutations in prostate cancer. |
We also review data regarding therapeutic options such as PARP inhibitors and PARP inhibitors in combination with novel hormonal agents. |
We discuss the encouraging results of recent PARP inhibitor or PARP inhibitor combinations and approvals, and call for an increase in clinical use of HRR gene mutation testing to inform and refine future efforts in the personalized treatment of prostate cancer. |
Introduction
Globally, prostate cancer (PC) is the second most common cancer in men and the fourth most common cancer overall [1]. In 2020, there were over 1.4 million new cases of PC worldwide, accounting for over 7% of all new cancer cases [1]. Early-stage PC is curable, with death from comorbidities often more likely to occur than death from PC [2], and a 10-year overall survival (OS) rate of 74% [3]. In contrast, long-term survival is poor among patients with metastatic castration-resistant PC (mCRPC), an advanced form of PC that does not respond to hormone therapy [2,3,4,5,6]. The median survival of patients with mCRPC is approximately 13 months after disease onset [5], and the 10-year OS rate is only 7% [6].
Surgery and radiation therapy remain the treatments of choice for patients with localized PC and can be curative. For patients with mCRPC, androgen deprivation therapy (ADT) and chemotherapy are among the most commonly used therapies [4]. However, recent improvements in the understanding of PC have informed the identification and development of novel treatment targets and strategies for patients with advanced disease. For example, interrogation of the PC metabolome has revealed potential metabolic treatment targets including enzymes involved in lipogenic pathways [7]. Furthermore, advances in high-throughput sequencing and the understanding of the molecular drivers of PC led to the development of targeted therapies that provide new options for patients with mCRPC [8]. Up to one-third of patients with mCRPC have homologous recombination repair pathway gene mutations (HRRm), including breast cancer associated genes 1/2 (BRCA1/BRCA2) and ataxia-telangiectasia mutated (ATM) [9]. PC with HRRm responds favorably to therapies that target homologous recombination repair (HRR), such as poly(ADP-ribose) polymerase (PARP) inhibitors [10]. Therefore, HRRm testing may improve the survival of patients with advanced or metastatic PC by hel** clinicians identify patients who could benefit from PARP inhibition. Nonetheless, the rate of HRRm testing in patients with PC is low [9]. Physicians have limited access to genomic testing and the lack of established and widely accepted methods for detection of HRRm in prostate biopsy specimens are important challenges limiting broader uptake of HRRm testing in patients with PC [9, 11].
Here, we comprehensively review the current knowledge regarding the prevalence and clinical significance of HRRm in PC. We also summarize the efficacy and mechanism of action of PARP inhibitors in the treatment of mCRPC with HRRm. This comprehensive overview aims to increase the adoption of HRRm testing, inform future efforts in personalized treatment of PC, and improve outcomes for patients with PC.
Methods
For this narrative review article, we searched PubMed with the following search terms: prostate cancer, homologous recombination repair, PARP inhibitors, immune checkpoint inhibitors. We then reviewed the titles and abstracts of the search results (including from congress abstracts) to identify relevant results. Additionally, we selected relevant trials from the ClinicalTrials.gov database (https://www.clinicaltrials.gov/) and assessed eligible recommendations and treatment guidelines from relevant associations. We also consulted the bibliographies of the selected articles to include all relevant studies. This narrative review article does not contain any new studies with human participants or animals.
Role of the HRR Pathway in PC
DNA Damage Repair via the HRR Pathway
The most common types of DNA damage are double-strand breaks (DSBs), single-strand breaks (SSBs), mismatches, alkylation, insertions, deletions, and introduction of bulky DNA adducts [12, 13]. DNA damage response (DDR) mechanisms vary depending on the type of damage. HRR is the primary mechanism by which cells accurately repair DSBs, using a homologous DNA template, and is highly regulated by multiple tumor suppressor proteins and complexes encoded by genes including BRCA1, BRCA2, radiation-sensitive protein 51 (RAD51), and partner and localizer of BRCA2 (PALB2) which are necessary for successful HRR [14,15,16]; the process has been described in detail elsewhere [16,17,18].
DNA damage repair is vital for the maintenance of genomic integrity. Deficiencies in HRR and other DNA repair mechanisms play an important role in the development of various cancers, including PC [19,20,21]. Deleterious HRRm (e.g., loss of BRCA1/BRCA2) can lead to deficiencies in DNA repair via the HRR pathway, which, in turn, can lead to accumulation of DNA damage and carcinogenesis [12]. Consistent with the potential role of HRR genes in prostate carcinogenesis, results from a large retrospective study of 1072 patients who underwent testing for BRCA1/BRCA2 mutations indicated that the incidence of PC was significantly higher in patients with BRCA2 mutations than in those without (incidence ratio, 4.9 [95% CI, 2.0–10.1]; P = 0.002) [22].
Alterations in DNA damage repair mechanisms also contribute to disease progression [23]. HRRm in PC has been associated with increased risk of recurrence, metastasis, and PC-specific death [24,25,26,27,28,29]. Accumulation of mutations in advanced tumors can result in increased genomic instability and mutational divergence, leading to treatment resistance, metastasis, and tumor recurrence [30]. Patients with HRRm often have a poor prognosis [31] and should be closely monitored by their health care team.
The androgen receptor (AR) pathway interacts with various DDR pathways, including HRR [32,33,34]. AR splice variants were found to promote PC cell survival after irradiation, and inhibition of AR splice variant interaction with DNA-dependent protein kinase (DNA-PK) increased DNA damage and PC cell death after irradiation [35]. AR splicing variant 7, one of the most abundant AR splice variants, enhanced DDR by activating PARP1 [36]. These findings suggest that AR splice variants induced by ADT may promote DDR and resistance to DNA damage-inducing therapies. These findings also indicate that combining DNA-PK or PARP inhibitors with ADT and radiotherapy may radiosensitize PC cells and improve treatment response [33, 34, 37].
In another study in vitro, treatment of castration-resistant PC cells with the AR pathway inhibitor enzalutamide suppressed the expression of various HRR genes, including BRCA1, RAD54L, and RMI2 [38]. Pretreatment of PC cells and mice bearing prostate tumors with enzalutamide enhanced the anticancer and proapoptotic effects of the PARP inhibitor olaparib, suggesting a potential synergistic effect [38]. Moreover, combined inhibition of the AR and checkpoint kinase 1 (CHK1) pathways promoted PC cell death, regardless of p53 status [39]. This crosstalk between DDR and AR signaling may contribute to the association between DDR and aggressive phenotypes in PC [21].
Prevalence of Germline and Somatic HRRm in PC
Genomic profiling studies have shown that HRRm is a critical oncogenic driver of PC and may predispose to PC.
A pan-tumor meta-analysis found that germline BRCA1 and BRCA2 mutations were in 0.5% and 3.5% of all PC, respectively, with somatic BRCA1 and BRCA2 mutations found in 5.7% and 3.2%, respectively [40]. This prevalence may increase in mCRPC: up to one-third of patients with mCRPC harbor HRRm, with approximately 23% of patients having non-BRCA1/BRCA2 HRRm [9], comparable to global clinical trials and genomic profiling studies reporting slightly over one-quarter of patients with mCRPC having an HRRm [11, 41,42,43].
Targeted sequencing of 139 cancer-related genes in 24 patients under the age of 63 years with PC revealed a total of 62 germline mutations in 45 genes; 22/24 patients harbored germline mutations, and nearly 60% of patients had mutations in DDR genes. BRCA2 (20.8%) and gap junction beta-2 (GJB2) (20.8%) were the most frequently mutated genes [44]. Mutations in CHEK2, BRCA1, PALB2, cyclin-dependent kinase inhibitor 2A (CDKN2A), homeobox protein B13 (HOXB13), protein phosphatase 1D (PPM1D), and ATP-dependent DNA helicase Q1 (RECQL) were also common (8.3% each) [44]. Another targeted sequencing study of 18 DDR genes in 316 patients with PC showed that 9.8% of patients had pathogenic germline mutations in 18 DDR genes [45]. BRCA2 was the most frequently mutated gene (germline mutations in 6.3% of patients), followed by BRCA1 and ATM (0.6% each). A whole-exome sequencing study showed that 31% of patients harbored deleterious germline mutations in DDR genes. Nearly 12% of patients harbored HRRm, 2.4% in mismatch repair (MMR) genes, and 16.7% in other DDR pathways [46]. Again, BRCA2 was the most commonly mutated HRR gene (5.3%).
Germline HRRm has also been identified in patients with metastatic PC. A sequencing study of 692 patients with metastatic PC revealed that 82 (11.8%) had a total of 84 germline mutations in 16 DDR genes [47]. As before, BRCA2 was the most commonly mutated gene (5.3%), followed by ATM (1.6%), CHEK2 (1.9%), BRCA1 (0.9%), and RAD51D (0.4%). Consistently, among patients with mCRPC treated with standard-of-care radium-223 in the prospective observational study PRORADIUM (NCT02925702), pathogenic germline HRRm was identified in 15/169 (8.8%) patients [48]. Germline mutations in BRCA2 were the most common (5/169 patients), followed by ATM (n = 4), BRCA1 (n = 1), BRCA1 + CHEK2 (n = 1), BRCA1 interacting protein 1 (BRIP1) (n = 1), nibrin (NBN) (n = 1), and Bloom syndrome protein (BLM) (n = 1) [48, 49].
HRRm Testing in PC
Guidelines on Germline or Somatic HRRm Testing in Patients with Newly Diagnosed PC
National Comprehensive Cancer Network (NCCN) guidelines for PC recommend germline testing for patients with newly diagnosed PC and a family history of high-risk germline mutations (e.g., BRCA1/BRCA2 mutation, Lynch syndrome), suspected family history, Ashkenazi Jewish ancestry, or presence of intraductal carcinoma on biopsy [50]. Germline mutation testing in these patients is recommended to include MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MSH6, and mismatch repair endonuclease PMS2 (PMS2; for Lynch syndrome) and the HRR genes BRCA1, BRCA2, ATM, PALB2, CHEK1, and CHEK2. Testing for HOXB13 should also be considered [50].
Similarly, European Association of Urology (EAU) guidelines recommend consideration of genetic testing for germline mutations in DDR genes (BRCA1, BRCA2, ATM, and MMR genes) in metastatic PC; men with a family history of high-risk germline mutations or a family history of multiple cancers on the same side of the family; men with high-risk PC and a family member diagnosed with PC at age < 60 years; and men with multiple family members diagnosed with clinically significant PC at age < 60 years or a family member who died from PC [51]. The EAU guidelines also recommend testing for somatic HRRm in patients diagnosed with metastatic PC.
Similar to the NCCN and EAU guidelines, European Society for Medical Oncology (ESMO) Clinical Practice Guidelines testing for germline mutations in BRCA1, BRCA2, and other DDR genes associated with cancer predisposition in all patients with metastatic PC and in those with a family history of cancer [52]. They also recommend testing for somatic mutations in HRR and MMR genes in patients with mCRPC and that patients with pathogenic somatic mutations should be referred for germline testing and genetic counseling [52].
Approved Methods for Detecting HRRm in PC
Testing for germline or somatic HRRm in patients with PC is mostly based on next-generation sequencing (NGS) of samples from metastatic lesions, primary tumors, or both. Testing for HRRm should be performed by an accredited institution using a standard NGS procedure and a minimum depth of coverage of 200× [51]. While we focus on approved methods here, considering that cost may limit the accessibility of testing, we note that there are many other HRRm testing methods based on NGS potentially used in clinical practice and that testing methods vary across studies.
The FoundationOne CDx Test is an NGS-based assay performed on DNA from solid tumor tissue. This assay has been approved by the US Food and Drug Administration (FDA) and is used in clinical trials to identify somatic HRRm in patients with PC [53, 54]. More recently, assays based on different samples or methods have been developed. For example, subgroup analysis of patients enrolled in the TRITON2 trial showed that patients with BRCA1/BRCA2-mutated mCRPC benefited from treatment with rucaparib, regardless of the sample used to assess BRCA1/BRCA2 mutation status (central plasma, central tissue, or local testing) [55].
Circulating tumor DNA (ctDNA) testing has also been used in some studies. FoundationOne Liquid CDx is an FDA-approved liquid biopsy NGS companion diagnostic test to assess HRRm status using ctDNA [56].
Prevalence of Homologous Recombination Deficiency in PC
Deleterious mutations in HRR genes (e.g., BRCA1/BRCA2) can lead to a phenotype of homologous recombination deficiency (HRD). As tumor cells with HRD are unable to accurately repair DNA damage using the HRR pathway, DNA damage accumulates and genomic instability increases [57, 58]. The genomic instability score in tumor tissue can be measured using algorithms that incorporate information regarding large-scale state transitions (LST), loss of heterozygosity (LOH), and telomeric allelic imbalance (TAI) [40, 57]. Although there is no consensus on the definition of HRD in PC, testing for HRD commonly involves testing for the presence of deleterious or suspected deleterious BRCA1/BRCA2 mutations or presence of genomic instability to generate a genomic instability score [40]. Genomic signatures have also been used to predict HRD status in tumor tissues, with methods in PC commonly relying on germline BRCA1/BRCA2 and HRR gene panel mutation testing [57]. However, definitions and assays used to determine HRD vary across studies and there is no standard clinical cutoff for genomic instability scores in PC.
Therefore, data on the prevalence of HRD in patients with PC remain limited [40]. An analysis of the HRD score (defined as the sum of LOH, LST, and TAI) in three cohorts of patients with primary PC (557 patients in total) showed that tumors with germline BRCA2 mutations had greater HRD scores than those with germline ATM or CHEK2 mutations (median HRD score, 27.0 vs. 16.5 [P = 0.029] and 9.0 [P < 0.001], respectively) [59]. Another analysis of molecular signatures in PC showed that tumor samples with mutations in BRCA1 or BRCA2, as well as a subset without mutations in BRCA1/BRCA2 genes, exhibited somatic HRD-associated mutation signatures [60]. These findings suggest that BRCA1/BRCA2 mutations in patients with PC are associated with the highest HRD scores and that testing for germline or somatic BRCA1/BRCA2 mutations may be a feasible method to identify patients who are most likely to respond to PARP inhibition.
Prognostic and Predictive Significance of HRRm in PC
Tumor Aggressiveness, Risk of Metastasis, and Patient Outcomes
Mutations in DNA repair genes may predict metastasis in PC. The exome-sequencing analysis by Armenia et al. [61] showed that mutations in genes involved in DNA repair, epigenetic regulation, PI3K signaling, cell cycle, and rat sarcoma (RAS)/rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase (MAPK) signaling were significantly more common in metastatic than primary tumors [61]. Consistently, a retrospective analysis of 150 patients with recurrent or metastatic PC suggested that the 21 (14%) patients with germline mutations in DDR genes were more likely to show intraductal/ductal histology (48% vs. 12%, P < 0.01) and lymphovascular invasion (52% vs. 14%, P < 0.01) than patients without DDR gene mutations [62]. Another analysis that included 2019 patients with PC indicated that germline mutations in BRCA1 or BRCA2 were significantly associated with increased tumor aggressiveness, including nodal involvement (P = 0.00005), metastasis at diagnosis (P = 0.005), T3/T4 stage (P = 0.003), and Gleason score ≥ 8 (P = 0.00003) [24]. Compared with patients without BRCA1/BRCA2 mutations, those with germline mutations in BRCA1 or BRCA2 had significantly longer median cause-specific survival (median, 15.7 vs. 8.6 years; hazard ratio [HR], 1.8; P = 0.015), a higher 5-year cause-specific survival rate (96.0% vs. 82.0%; HR, 2.6; P = 0.01), and a higher 5-year metastasis-free survival rate (93.0% vs. 77.0%; HR, 2.7; P = 0.009) [24]. Testing for germline and somatic mutations may also be useful for initial risk stratification of patients with newly diagnosed early-stage PC. Currently, molecular testing is recommended for selected patients with PC during active surveillance or after radical prostatectomy, as mutations in DDR and cell-cycle genes can predict the risk of metastasis, biochemical recurrence, and cancer-specific death [50].
Cyclin dependent kinase 12 (CDK12) is implicated in DNA repair, cell cycle, RNA splicing, and cell differentiation [63,64,65]. CDK12 loss in PC cells results in genomic instability, characterized by a high number of gene fusions [63, 64]. A retrospective multicenter study in PC showed that loss-of-function (LOF) mutations in CDK12 were associated with aggressive tumor phenotypes, including Gleason grade 4 or 5 and metastases at diagnosis [66]. Another study of 913 biopsies from patients with PC indicated that patients with mCRPC harboring biallelic CDK12 mutations had significantly shorter OS than patients without CDK12 mutations (median, 5.1 [95% CI, 4.0–7.9] vs. 6.4 years [95% CI, 5.7–7.8]; HR, 1.65 [95% CI, 1.07–2.53]; P = 0.02), suggesting a potential prognostic role of CDK12 mutations in PC, although this was not confirmed with multivariate analysis [67].
Treatment Response
Radiopharmaceuticals
Even though the clinical adoption of HRRm testing for in PC remains low, it has been hypothesized that HRRm may predict survival outcomes and response to radiopharmaceutical treatment with radium-223. Analysis of data from the prospective observational PRORADIUM study in patients with mCRPC with or without a germline HRRm showed that pathogenic germline HRRm was associated with improved alkaline phosphatase (ALP) responses after standard-of-care radium-223 [48, 49]. Twelve weeks after treatment, a > 30% decline in ALP was observed in 71.4% of patients with germline HRRm and in 39.5% of patients without HRRm (P = 0.022). However, no significant differences in 12-week prostate-specific antigen (PSA) decline (18% in both groups) median radiographic (r) progression-free survival (rPFS, 5.6 vs. 6.3 months; P = 0.063) or median OS (16.8 vs. 11.5 months, P = 0.078) were observed between the two groups [48, 49].
PARP Inhibitors
LOF alterations in DDR genes, including HRR genes, are associated with response to PARP inhibition in prostate and other cancers. Data in PC suggest that germline or somatic mutations in BRCA1/BRCA2 are associated with improved response to PARP inhibitors, including olaparib, niraparib, and rucaparib [41, 68,69,70,71]. An analysis of HRD scores in patients with primary PC revealed that the highest HRD scores were found among patients with germline BRCA2 mutations [59]. While mean PSA reductions among patients who received olaparib were similar between HRD groups of ≥ 25 or < 25, median PFS was longer among patients with higher HRD scores (14.9 months [range, 10.1–19.8] vs. 9.9 months [range, 6.8–11.0]) [59]. Larger studies are warranted to confirm the ability of HRD scores to predict response to PARP inhibition in mCRPC.
ATM regulates DNA damage repair by sensing DNA damage, preventing cell-cycle progression, and initiating DDR pathways [72]. Recent data suggest that ATM loss in PC may predict response to PARP inhibitors in combination with ataxia telangiectasia and Rad3-related protein (ATR) inhibitors, consistent with preclinical work [73]. PC cells with ATM loss exhibit high levels of genomic instability, which might explain increased sensitivity to combined treatment with PARP and ATR inhibitors [73].
A retrospective multicenter study showed that among patients with LOF mutations in CDK12 who underwent first-line ADT for metastatic hormone-sensitive PC (n = 59), the PSA response rate was 79.7%, and the median PFS was 12.3 months (95% CI, 9.1–17.0) [66]. Among patients who received first-line abiraterone and enzalutamide for mCRPC (n = 34), the PSA response rate was 41.2%, and the median PFS was 5.3 months (95% CI, 3.7–6.9) [66]. Of the 11 patients who received PARP inhibitors (olaparib [n = 10] or rucaparib [n = 1]), none had a PSA response; the median PFS was 3.6 months (95% CI, 3.0–4.2) [66].
PALB2 and BRCA1 associated RING domain 1 (BARD1) are essential HRR genes that regulate DSB repair. In HRR, PALB2 acts as a bridging molecule, facilitating the formation of a BRCA complex consisting of BRCA1, BRCA2, and RAD51 [74]; BARD1 also binds to BRCA1 to coordinate activity via E3 ubiquitin ligase activity [75]. Mutations in PALB2 and BARD1 have been shown to be associated with PARP inhibitor benefit in PC cells [75] and PALB2 mutations have in the TOPARP-B trial [76]. The potential benefits of PARP inhibitors alone or in combination with chemotherapy in patients with PALB2-mutant PC require confirmation in future randomized clinical trials.
Immune Checkpoint Inhibitors
HRRm can lead to increased tumor mutational burden, which can influence immunotherapy response. Emerging evidence suggests that mutations in some HRR genes may predict response to immunotherapy in patients with PC. Among nine patients with PC who had LOF mutations in CDK12 and were treated with a programmed cell death 1 (PD-1) inhibitor (pembrolizumab [n = 5] or nivolumab [n = 4]) in a multicenter study, the PSA response rate was 33.3%, and the median PFS was 5.4 months (95% CI, 3.2–10.8) [66]. In a large study of 913 biopsies from patients with PC, the proportion of immunosuppressive cluster of differentiation 4 (CD4)+ forkhead box protein 3 (FOXP3)− cells was significantly higher in patients with PC harboring CDK12 mutations than in those without CDK12 mutations (50.5 vs. 6.2 cells/mm2; P < 0.0001) [67]. The higher levels of immunosuppressive immune cells infiltrating the tumor may explain the low response rates to immune checkpoint inhibition observed in patients with PC harboring CDK12 mutations. The findings of van Wilpe et al. [77] support the view that PC with HRD should be considered an immunologically distinct subtype characterized by an altered peripheral T cell receptor repertoire. Although these findings suggest that CDK12 loss is associated with limited susceptibility to immune checkpoint inhibition in patients with PC, larger prospective clinical trials are required to determine the clinical value of immunotherapy in patients with CDK12-mutant PC.
Role of HRRm in Clinical Decision-Making for Patients with PC
PARP inhibitors
Rationale for PARP Inhibitors with PC Harboring HRRm
Various PARP inhibitors have been approved for a subset of patients with prostate, ovarian, breast, and pancreatic cancer [78, 79]. Biallelic HRRm results in dysfunctional HRR proteins and HRD. Hence, tumors with HRRm are particularly sensitive to agents that induce DNA damage [78, 79]. Members of the PARP family are part of the complex that mediates the recognition and repair of DNA damage via the base excision repair pathway [80, 81]. In cells with HRD, base excision repair is the main pathway for DNA repair, especially for DSBs. When combined with cytotoxic chemotherapy, PARP inhibition in tumor cells with HRD can result in the accumulation of DSBs and, ultimately, cell death—a mechanism termed synthetic lethality. Various preclinical studies have confirmed the strong and selective cytotoxic activity of PARP inhibitors in cancer cells with mutations in HRR genes, including BRCA1 and BRCA2 [82, 83].
Efficacy of Approved PARP Inhibitors in PC Harboring HRRm
In 2020, the FDA approved two PARP inhibitors, olaparib and rucaparib, for use as monotherapy in patients with mCRPC harboring germline or somatic aberrations in BRCA1/BRCA2 or non-BRCA1/BRCA2 HRR genes (olaparib) or BRCA1/BRCA2 (rucaparib) [84, 85], with olaparib gaining subsequent European Medicines Agency (EMA) approval in the same setting [86]. In China, olaparib was granted approval for BRCA1/BRCA2-mutated mCRPC. These approvals were based on the promising antitumor activity of these agents in the pivotal clinical trials PROfound and TRITON2 (Table 1). Trials are also ongoing in metastatic castration-sensitive PC (Table 2).
PROfound was a randomized phase 3 trial investigating the use of the PARP inhibitor olaparib in men with mCRPC with progression on previous hormonal therapy [41]. Tumors in all patients harbored LOF alterations in at least one HRR gene, including ATM, BRCA1, BRCA2, BRIP1, BARD1, CDK12, CHEK1, CHEK2, PALB2, PPP2R2A, RAD51B, RAD51C, and RAD51D. Patients were randomly assigned to olaparib or the physician’s choice of control treatment (enzalutamide or abiraterone). Among the 245 patients with mutations in BRCA1, BRCA2, or ATM, median PFS was significantly longer in the olaparib than control group (7.4 vs. 3.6 months; HR, 0.34; 95% CI, 0.25–0.47; P < 0.001) [41]. Compared with the control group, the olaparib group had a significantly higher objective response rate (ORR; 33% [28 of 84] vs. 2% [1 of 43]; odds ratio, 20.86 [95% CI, 4.18–379.18]; P < 0.001), longer time to pain progression (HR, 0.44; 95% CI, 0.22–0.91; P = 0.02), and final analysis confirmed a longer median OS (19.1 vs. 14.7 months; HR, 0.69; 95% CI, 0.50–0.97; P = 0.02) [41, 87]. These results suggest that olaparib represents a promising treatment option for patients with mCRPC harboring HRRm and with disease progression on enzalutamide or abiraterone.
TRITON2 was a randomized phase 2 trial investigating rucaparib in 277 men with mCRPC who had progressed after one or two lines of previous hormonal therapy and one taxane-based chemotherapy [70, 88]. All patients had deleterious tumor mutations in BRCA1, BRCA2, ATM, or another HRR gene. Among patients with a BRCA1/BRCA2 mutation (n = 81), the confirmed ORR (independent radiology review) was 46% (95% CI, 35–57), and the PSA response rate (≥ 50% decrease from baseline) was 53% (95% CI, 46–61). Compared with patients with BRCA1 mutations, those with BRCA2 mutations had a higher PSA response rate, although the ORR was similar. Among BRCA1/BRCA2-mutated patients, the median PFS based on radiologic assessment was 10.7 months (95% CI, 8.7–13 months). Anemia was the most common grade ≥ 3 treatment-emergent adverse event (TEAE; 29%).
Subsequent analysis of data the TRITON2 trial showed that the method of BRCA1 or BRCA2 mutation testing (central plasma, central tissue, or local testing) did not significantly affect response to rucaparib, as PSA response rates and ORRs were similar in the three patient subgroups [55]. Similarly, plasma testing in the PROfound trial found that patients with ctDNA mutations in BRCA1/BRCA2 or ATM had a similar benefit in PFS from olaparib to those who had BRCA1/BRCA2 or ATM mutations in tumor tissue [89]. These findings suggest that plasma testing may be a convenient method for identifying men with mCRPC who may benefit most from PARP inhibition.
Other Trials of PARP Inhibitors in mCRPC
The safety and efficacy of several PARP inhibitors are currently being evaluated in patients with mCRPC harboring HRRm (Table 1). TALAPRO-1 (NCT03148795) is a multicenter phase 2 study evaluating the use of the PARP inhibitor talazoparib in men with mCRPC who progressed on prior ADT (enzalutamide, abiraterone, or both) and taxane-based chemotherapy [90]. All patients had monoallelic or biallelic alterations in DDR genes involved directly or indirectly in HRR, including BRCA1, BRCA2, ATM, ATR, CHEK2, Fanconi anemia complementation group A (FANCA), MRE11A, PALB2, or RAD51C. After a median follow-up of 16.4 months (interquartile range [IQR], 11.1–22.1 months), the ORR was 29.8% (95% CI, 21.2–39.6), the PSA response rate (≥ 50% reduction) was 42%, and the median rPFS was 5.6 months (95% CI, 3.7–8.8). Anemia (31%), thrombocytopenia (9%), and neutropenia (8%) were the most frequent grade 3–4 TEAEs in the safety analysis population (n = 127).
GALAHAD (NCT0285443) was a multicenter phase 2 study evaluating the PARP inhibitor niraparib in patients with mCRPC with progression on prior ADT and taxane-based chemotherapy [71] and who had germline or somatic biallelic pathogenic alterations in BRCA1 or BRCA2. At a median follow-up of 10 months, the ORR was 34.2% (95% CI, 23.7–46.0). The median rPFS was 8.08 months (95% CI, 5.55–8.38), the median OS was 13.01 months (95% CI, 11.04–14.29), and the PSA response rate was 43% (95% CI, 34.7%–51.5%). Nausea (58%), anemia (54%), and vomiting (38%) were the most common any-grade TEAEs in the safety analysis population (n = 289). Anemia (33%), thrombocytopenia (16%), and neutropenia (10%) were the most frequent grade 3–4 TEAEs. Two deaths were deemed likely related to niraparib treatment [71].
TRAP (NCT03787680) is a phase 2 study investigating olaparib in patients with mCRPC [91, 92]. Patients with tumors that progressed after at least one more line of treatment were enrolled and treated with olaparib in combination with ceralasertib (AZD6738), an ATR inhibitor [91]. At data cutoff (DCO), the rate of confirmed ≥ 50% PSA decline was 33% (4/12) in patients who tested positive for DNA repair deficiency (defined as germline or somatic BRCA2 or ATM loss) and 11% (4/35) in patients who tested negative for DNA repair deficiency [91]. BRCA2 mutations were associated with treatment response. More mature data and subgroup analyses are needed to confirm the clinical benefits of olaparib plus ceralasertib in patients with mCRPC harboring HRRm.
TRITON3 (NCT02975934) is a multicenter phase 3 randomized controlled trial (RCT) evaluating the efficacy of rucaparib monotherapy in patients with mCRPC harboring deleterious germline or somatic mutations in BRCA1, BRCA2, or ATM [93]. Patients were randomized to receive rucaparib or the physician’s choice of therapy (abiraterone, enzalutamide, or docetaxel). Similar to previous studies of PARP inhibitors in this setting, median PFS was significantly longer in the rucaparib arm (10.2 months) than the control arm (6.4 months), with a hazard ratio of 0.61 (95% CI, 0.47–0.80), although this benefit did not appear to extend to patients with an ATM mutation (median PFS, 8.1 vs. 6.8 months; HR, 0.95 [95% CI, 0.59–1.52) [94].
Combination of PARP Inhibitors with Novel Hormonal Agents
Rationale for Combining Novel Hormonal Agents with PARP Inhibitors in PC
Novel hormonal agents (NHA), such as abiraterone and enzalutamide, have been approved for the treatment of patients with mCRPC who have progressed on prior therapies. The potential use of abiraterone and enzalutamide as first-line treatment for PC and their combination with PARP inhibitors have also been explored (Fig. 1). The combined use of NHA and PARP inhibitors is based on recent in vitro evidence of the interplay between the AR signaling pathway and various DDR pathways [33, 34, 37], and PARP inhibition may therefore prevent AR-mediated DNA repair [35, 36]. Preclinical evidence also suggests that enzalutamide treatment may suppress the expression of various HRR genes in castration-resistant PC cells [38, 39] and that AR inhibition is associated with increased PARP activity [95], providing a strong rationale for combining PARP inhibitors with NHAs and radiotherapy in patients with PC harboring HRRm.

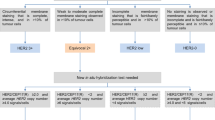
Mechanisms of PARP and AR interactions that lead to synergistic PARP inhibitor and NHA benefit in mCRPC. Left: DNA damage in the form of SSBs or DSBs, in the presence of PARP and AR inhibition, lead to DSB accumulation and impaired DNA repair. This subsequently leads to lethal genomic instability and prostate cancer cell death. Right: PARP and AR interactions in prostate cancer. PARP is critical for BER of SSBs; inhibition of PARP leads to the conversion of SSBs to DSBs, which require functional HRR for accurate repair. Purple: PARP can promote AR target gene transcription and play a key role in the occurrence and development of prostate cancer. PARP interacts with AR via transcriptional co-regulators and therefore augments AR activity by regulating gene transcription. Gold: Activation of AR signaling can affect DNA repair mechanisms and the overall DNA damage response of prostate cancer cells. AR has multiple target genes and include those implicated in multiple DNA repair pathways, including multiple genes that are important for HRR. In addition, inhibition of AR signaling is associated with increased PARP activity. AR androgen receptor; BER base excision repair; DSB double-stranded break; FANC Fanconi anemia; HRR homologous recombination repair; MMR mismatch repair; MRN MRe11, Rad50, and Nbs1 complex; NHA novel hormonal agent; NHEJ non-homologous end joining; PARP poly(ADP) ribose polymerase; SSB single-stranded break
Efficacy and Ongoing Trials of Novel Hormonal Agents Combined with PARP Inhibitors in PC
Several trials have investigated the combination of PARP inhibition and NHA [42, 96,97,98]. Study 08 was a phase 2 RCT investigating the use of abiraterone plus olaparib in 142 patients with mCRPC who were previously treated with docetaxel and up to one additional line of chemotherapy. Abiraterone plus olaparib was superior to abiraterone plus placebo in improving rPFS (median, 13.8 [95% CI, 10.8–20.4] vs. 8.2 months [95% CI, 5.5–9.7]; HR, 0.65 [95% CI, 0.44–0.97]; P = 0.034) [96]. Median time to second progression or death (23.3 vs. 18.5 months; HR, 0.79 [95% CI, 0.51–1.21]; P = 0.28) and median OS (22.7 vs. 20.9 months; HR, 0.91 [95% CI, 0.60–1.38]; P = 0.66) were also prolonged with combined treatment compared with abiraterone alone; however, these improvements were not statistically significant [96]. A prespecified exploratory analysis of data from Study 08 showed no significant differences in pain or health-related quality of life between patients treated with abiraterone plus olaparib and those who received abiraterone plus placebo [99]. The incidence of serious adverse events was higher in patients who received abiraterone plus olaparib than in those treated with abiraterone plus placebo [96].
The phase 3 randomized PROpel trial in previously untreated patients with mCRPC showed that first-line abiraterone plus olaparib was superior to abiraterone plus placebo in prolonging imaging-based PFS (median, 24.8 vs. 16.6 months; HR, 0.66 [95% CI, 0.54–0.81]; P < 0.001) [42]. A later, prespecified analysis of OS (47.9% data maturity) found a clinically meaningful benefit (median, 42.1 vs. 34.7 months; HR, 0.81 [95% CI, 0.67–1.00]; P = 0.054) [100]. Notably, olaparib plus abiraterone prolonged PFS in patients with mCRPC irrespective of HRRm status [42], with the greatest benefit in OS seen in the BRCA1/BRCA2-mutated subgroup (HR, 0.29 [95% CI, 0.14–0.56]) [100]. The combination had a good tolerability profile; the most frequent adverse events were anemia, fatigue, and nausea [42]. Similarly, the phase 3 randomized TALAPRO-2 study evaluated first-line talazoparib plus enzalutamide versus placebo plus enzalutamide in patients with mCRPC with or without HRRm [97]. The primary endpoint of rPFS was superior in the talazoparib plus enzalutamide arm (median, not reached vs. 21.9 months; HR, 0.63 [95% CI, 0.51–0.78]) and subgroup analysis showed the greatest benefit in patients with HRRm (27.9 vs. 16.4 months; HR, 0.46 [0.30–0.70]) [97]. The benefit observed in these phase 3 RCTs swiftly led to FDA approvals for olaparib plus abiraterone and prednisone in BRCA1/BRCA2-mutated mCRPC (PROpel) and talazoparib plus enzalutamide in HRRm mCRPC (TALAPRO-2) [101, 102].
MAGNITUDE was a phase 3 RCT investigating the combination of niraparib plus abiraterone and prednisone vs. abiraterone and prednisone plus placebo in previously untreated patients with mCRPC, with or without HRRm (ATM, BRCA1, BRCA2, BRIP1, CDK12, CHEK2, FANCA, histone deacetylase 2 (HDAC2), PALB2) [103]. At a median follow-up of 18.6 months, median rPFS in patients with HRRm was 16.5 months in the combination therapy group and 13.7 months in the placebo group (HR, 0.73 [95% CI, 0.56–0.96]; P = 0.0217) [104]. Median OS in patients with HRRm was 16.5 months in the combination therapy group and 13.7 months in the placebo group (HR, 0.94 [95% CI, 0.65–1.36]; P = 0.7333). Additionally, combination therapy significantly improved ORR and delayed time to initiation of cytotoxic chemotherapy, time to symptomatic progression, and time to PSA progression in patients with HRRm [104]. A subsequent subgroup analysis with an additional 8 months of follow-up revealed the greatest benefit in rPFS was in patients with a BRCA1/BRCA2 mutation (HR, 0.55 [95% CI, 0.39–0.78]) [105]. Interestingly, while efficacy was observed regardless of HRR biomarker status in the PROpel and TALAPRO-2 studies of PARP inhibitor plus NHAs, with the greatest benefit among patients with an HRRm, no benefit was observed in patients without an HRRm in the MAGNITUDE study.
CASPAR (Alliance A031902; NCT04455750) is an ongoing phase 3 RCT investigating the efficacy of first-line enzalutamide combined with rucaparib or placebo in patients with mCRPC, regardless of HRRm status [106]. Preliminary results from CASPAR are expected in 2023. AMPLITUDE (NCT04497844) and TALAPRO-3 (NCT04821622) are ongoing phase 3 RCTs investigating the efficacy of niraparib or talazoparib in combination with enzalutamide or abiraterone plus prednisone in patients with metastatic castration-sensitive PC harboring deleterious germline or somatic mutations in HRR genes [107, 108].
Immune Checkpoint Inhibitors
Treatment with PARP inhibitors can lead to accumulation of DNA damage, and so may increase tumor mutational burden and enhance response to immunotherapy. However, there are limited clinical data on the efficacy of combined treatment with immunotherapy and PARP inhibitors in patients with PC harboring HRR gene mutations. In a phase 2 study, 17 patients with mCRPC with and without DDR gene mutations were treated with durvalumab plus olaparib after progression on neoadjuvant hormonal therapy (NCT02484404). The median rPFS for the entire cohort was 16.1 months (95% CI, 4.5–16.1 months), the 1-year rPFS rate was 51.5% (95% CI, 25.7–72.3%) [109], and the rate of radiographic or PSA response was 53% (9 of 17 patients). The median rPFS in patients with DDR gene mutations was 16.1 months (95% CI, 7.8–18.1 months). Notably, DDR gene alterations were associated with improved response to durvalumab plus olaparib. The PSA response rate was 100% (6 of 6 patients) in patients with biallelic BRCA2 mutations and 27% (3 of 11 patients) in patients without biallelic HRRm [109].
KEYNOTE-365 is an ongoing phase 1b/2 trial investigating the efficacy of pembrolizumab in combination with olaparib or other regimens in patients with mCRPC. Cohort A is investigating pembrolizumab plus olaparib. With a median time from allocation to DCO of 24 months (median duration of therapy, 5 months), the PSA response rate in patients treated with pembrolizumab plus olaparib was 15% (15 of 102 patients), ORR was 8% (5 of 59 patients), median PFS was 4.5 months (95% CI, 4.0–6.5 months), and median OS was 10 months (95% CI, 6.5–20) [110]. Cohort B investigated pembrolizumab plus docetaxel and prednisone. With a median time from allocation to DCO of 32 months, the PSA response rate was 34% (35 of 103 patients), ORR was 23% (12 of 52 patients), and median PFS was 8.5 months (95% CI, 8–10) [111].
KEYLYNK-010 was a phase 3 trial investigating the efficacy of pembrolizumab plus olaparib compared with abiraterone/enzalutamide in heavily pretreated patients with mCRPC [112]. At a planned interim analysis with a median follow-up of 11.9 months, the primary endpoints of PFS and OS were not significantly different between the two groups, despite a higher ORR in patients treated with pembrolizumab plus olaparib than in those treated with abiraterone or enzalutamide (17% vs. 6%; P = 0.002). Subgroup analysis showed that, in contrast to patients without HRRm, those with HRRm had improved PFS after treatment with pembrolizumab plus olaparib (HR, 0.53 [95% CI, 0.33–0.86] vs. 1.19 [95% CI, 0.90–1.58]); however, OS was similar in the two HRR subgroups [112].
Discussion and Conclusions
Evidence from clinical trials suggests that patients with metastatic PC harboring mutations in HRR genes, including BRCA1 and BRCA2, may benefit from treatment with DNA-damaging therapies and PARP inhibitors. The recent regulatory approvals of olaparib and rucaparib for use in patients with biomarker-positive mCRPC mark a new era in the treatment of mCRPC, for which treatment options remain limited. However, despite the role of HRRm in patients with mCRPC, the rate of molecular testing in patients with PC remains low [9]. Increasing awareness of the clinical significance of HRRm in patients with mCRPC, expanding physicians’ access to genomic testing, and reducing the costs of molecular diagnostic assays may help increase the use of HRRm testing in patients with PC and so help clinicians identify patients who are most likely to benefit from PARP inhibitors. This is particularly important for patients with mCRPC, for whom treatment options are limited. Reliable biomarkers for mCRPC are lacking, and expanding genetic testing for HRRm may help uncover valuable biomarkers that could improve risk stratification and the prediction of response to various treatments, including targeted therapy and immunotherapy.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed.
References
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Daskivich TJ, Fan KH, Koyama T, et al. Effect of age, tumor risk, and comorbidity on competing risks for survival in a U.S. population-based cohort of men with prostate cancer. Ann Intern Med. 2013;158(10):709–17.
Tamada S, Iguchi T, Kato M, et al. Time to progression to castration-resistant prostate cancer after commencing combined androgen blockade for advanced hormone-sensitive prostate cancer. Oncotarget. 2018;9(97):36966–74.
Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: a review. JAMA. 2017;317(24):2532–42.
Aly M, Leval A, Schain F,et al. Survival in patients diagnosed with castration-resistant prostate cancer: a population-based observational study in Sweden. Scand J Urol. 2020;54(2):115–21.
Tangen CM, Faulkner JR, Crawford ED, et al. Ten-year survival in patients with metastatic prostate cancer. Clin Prostate Cancer. 2003;2(1):41–5.
Lucarelli G, Loizzo D, Ferro M, et al. Metabolomic profiling for the identification of novel diagnostic markers and therapeutic targets in prostate cancer: an update. Expert Rev Mol Diagn. 2019;19(5):377–87.
Rubin MA, Demichelis F. The genomics of prostate cancer: emerging understanding with technologic advances. Mod Pathol. 2018;31(S1):S1–11.
Leith A, Ribbands A, Kim J, et al. Real-world homologous recombination repair mutation testing in metastatic castration-resistant prostate cancer in the USA, Europe and Japan. Future Oncol. 2022;18(8):937–51.
Marshall CH, Antonarakis ES. Therapeutic targeting of the DNA damage response in prostate cancer. Curr Opin Oncol. 2020;32(3):216–22.
Scott RJ, Mehta A, Macedo GS, et al. Genetic testing for homologous recombination repair (HRR) in metastatic castration-resistant prostate cancer (mCRPC): challenges and solutions. Oncotarget. 2021;12(16):1600–14.
Alhmoud JF, Woolley JF, Al Moustafa AE, Malki MI. DNA damage/repair management in cancers. Cancers (Basel). 2020;12(4):1050.
Wagener-Ryczek S, Merkelbach-Bruse S, Siemanowski J. Biomarkers for homologous recombination deficiency in cancer. J Pers Med. 2021;11(7):612.
Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21(10):3445–50.
Wang Y, Cortez D, Yazdi P, et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14(8):927–39.
Krajewska M, Fehrmann RS, de Vries EG, van Vugt MA. Regulators of homologous recombination repair as novel targets for cancer treatment. Front Genet. 2015;6:96.
Yang H, Jeffrey PD, Miller J, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297(5588):1837–48.
Pellegrini L, Yu DS, Lo T, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420(6913):287–93.
Mateo J, Boysen G, Barbieri CE, et al. DNA repair in prostate cancer: biology and clinical implications. Eur Urol. 2017;71(3):417–25.
Cimadamore A, Lopez-Beltran A, Massari F, et al. Germline and somatic mutations in prostate cancer: focus on defective DNA repair, PARP inhibitors and immunotherapy. Future Oncol. 2020;16(5):75–80.
Zhang W, van Gent DC, Incrocci L, van Weerden WM, Nonnekens J. Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate Cancer Prostatic Dis. 2020;23(1):24–37.
Mersch J, Jackson MA, Park M, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121(2):269–75.
Zhou J, Zhou XA, Zhang N, Wang J. Evolving insights: how DNA repair pathways impact cancer evolution. Cancer Biol Med. 2020;17(4):805–27.
Castro E, Goh C, Olmos D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31(14):1748–57.
Castro E, Goh C, Leongamornlert D, et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur Urol. 2015;68(2):186–93.
Marshall CH, Fu W, Wang H, et al. Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis. 2019;22(1):59–65.
Na R, Zheng SL, Han M, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71(5):740–7.
Castro E, Romero-Laorden N, Del Pozo A, et al. PROREPAIR-B: a prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2019;37(6):490–503.
Mateo J, Cheng HH, Beltran H, et al. Clinical outcome of prostate cancer patients with germline DNA repair mutations: retrospective analysis from an international study. Eur Urol. 2018;73(5):687–93.
Mitchell T, Neal DE. The genomic evolution of human prostate cancer. Br J Cancer. 2015;113(2):193–8.
Yip S, Khalaf D, Struss WJ, et al. Outcomes in patients (Pts) with advanced prostate cancer and inactivating germline mutations in BRCA2 or ATM. J Clin Oncol. 2018;36(6_suppl):242–242.
Guo X, Bai Y, Zhao M, et al. Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Res. 2018;46(2):689–703.
Polkinghorn WR, Parker JS, Lee MX, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3(11):1245–53.
Goodwin JF, Schiewer MJ, Dean JL, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3(11):1254–71.
Yin Y, Li R, Xu K, et al. Androgen receptor variants mediate DNA repair after prostate cancer irradiation. Cancer Res. 2017;77(18):4745–54.
Luo H, Liu Y, Li Y, et al. Androgen receptor splicing variant 7 (ARv7) promotes DNA damage response in prostate cancer cells. FASEB J. 2022;36(9):e22495.
Asim M, Tarish F, Zecchini HI, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8(1):374.
Li L, Karanika S, Yang G, et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal. 2017. https://doi.org/10.1126/scisignal.aam7479.
Karanika S, Karantanos T, Li L, et al. Targeting DNA damage response in prostate cancer by inhibiting androgen receptor-CDC6-ATR-Chk1 signaling. Cell Rep. 2017;18(8):1970–81.
Shao C, Wan J, Lam FC, et al. A comprehensive literature review and meta-analysis of the prevalence of pan-cancer BRCA mutations, homologous recombination repair gene mutations, and homologous recombination deficiencies. Environ Mol Mutagen. 2022;63(6):308–16.
de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091–102.
Clarke Noel W, Armstrong Andrew J, Thiery-Vuillemin A, et al. Abiraterone and olaparib for metastatic castration-resistant prostate cancer. NEJM Evid. 2022;1(9):EVIDoa2200043.
Abida W, Armenia J, Gopalan A, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol. 2017;2017:PO.17.00029.
Tang T, Tan X, Wang Z, et al. Germline mutations in patients with early-onset prostate cancer. Front Oncol. 2022;12:826778.
Wei Y, Wu J, Gu W, et al. Germline DNA repair gene mutation landscape in Chinese prostate cancer patients. Eur Urol. 2019;76(3):280–3.
Wu J, Wei Y, Pan J, et al. Prevalence of comprehensive DNA damage repair gene germline mutations in Chinese prostate cancer patients. Int J Cancer. 2021;148(3):673–81.
Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443–53.
Castro E, Mejorada RL, Saez M, et al. Impact of germline mutations in homologous recombination (HR) genes on the response to radium-223 for metastatic castration resistant prostate cancer (mCRPC). Ann Oncol. 2019;30:v343–4.
Castro E, Lozano Mejorada R, Medina A, et al. 590P PRORADIUM: prospective analysis of the impact of germline mutations in homologous recombination (HR) genes on the response to radium-223 for metastatic castration resistant prostate cancer (mCRPC). Ann Oncol. 2021;32:S638–9.
Mohler JL, Antonarakis ES, Armstrong AJ, et al. Prostate cancer, version 2.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2019;17(5):479–505.
EAU Guidelines on Prostate Cancer. Edn. presented at the EAU annual congress Amsterdam 2022. ISBN 978-94-92671-16-5.
Parker C, Castro E, Fizazi K, et al. Prostate cancer: ESMO Clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(9):1119–34.
Anscher MS, Chang E, Gao X, et al. FDA approval summary: rucaparib for the treatment of patients with deleterious BRCA-mutated metastatic castrate-resistant prostate cancer. Oncologist. 2021;26(2):139–46.
US Food & Drug Administration. FDA announces approval, CMS proposes coverage of first breakthrough-designated test to detect extensive number of cancer biomarkers. https://www.fda.gov/news-events/press-announcements/fda-announces-approval-cms-proposes-coverage-first-breakthrough-designated-test-detect-extensive#:~:text=The%20U.S.%20Food%20and%20Drug,in%20any%20solid%20tumor%20type. Accessed 2022 Aug 30.
Loehr A, Patnaik A, Campbell D, et al. Response to rucaparib in BRCA-mutant metastatic castration-resistant prostate cancer identified by genomic testing in the TRITON2 study. Clin Cancer Res. 2021;27(24):6677–86.
US Food & Drug Administration. FDA approves liquid biopsy NGS companion diagnostic test for multiple cancers and biomarkers. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-liquid-biopsy-ngs-companion-diagnostic-test-multiple-cancers-and-biomarkers#:~:text=FDA%20approves%20liquid%20biopsy%20NGS%20companion%20diagnostic%20test%20for%20multiple%20cancers%20and%20biomarkers,-Share&text=On%20October%2026%20and%20November,(Foundation%20Medicine%2C%20Inc.). Accessed 2022 Aug 30.
Stewart MD, Merino Vega D, Arend RC, et al. Homologous recombination deficiency: concepts, definitions, and assays. Oncologist. 2022;27(3):167–74.
Miller RE, Leary A, Scott CL, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;31(12):1606–22.
Lotan TL, Kaur HB, Salles DC, et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod Pathol. 2021;34(6):1185–93.
Sztupinszki Z, et al. Detection of molecular signatures of homologous recombination deficiency in prostate cancer with or without BRCA1/2 mutations. Clin Cancer Res. 2020;26(11):2673–80.
Armenia J, Diossy M, Krzystanek M, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50(5):645–51.
Isaacsson Velho P, Silberstein JL, Markowski MC, et al. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate. 2018;78(5):401–7.
Liang S, Hu L, Wu Z, et al. CDK12: a potent target and biomarker for human cancer therapy. Cells. 2020;9(6):1483.
Magnuson B, Bedi K, Narayanan IV, et al. CDK12 regulates co-transcriptional splicing and RNA turnover in human cells. iScience. 2022. https://doi.org/10.1016/j.isci.2022.105030.
Chirackal Manavalan AP, Pilarova K, Kluge M, et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep. 2019;20(9):e47592.
Antonarakis ES, Isaacsson Velho P, Fu W, et al. CDK12-altered prostate cancer: clinical features and therapeutic outcomes to standard systemic therapies, poly (ADP-ribose) polymerase inhibitors, and PD-1 inhibitors. JCO Precis Oncol. 2020;4:370–81.
Rescigno P, Gurel B, Pereira R, et al. Characterizing CDK12-mutated prostate cancers. Clin Cancer Res. 2021;27(2):566–74.
Boussios S, Rassy E, Moschetta M, et al. BRCA mutations in ovarian and prostate cancer: bench to bedside. Cancers (Basel). 2022;14(16):3888.
Marshall CH, Sokolova AO, McNatty AL, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol. 2019;76(4):452–8.
Abida W, Patnaik A, Campbell D, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. 2020;38(32):3763–72.
Smith MR, Scher HI, Sandhu S, et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2022;23(3):362–73.
Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5(9):a012716.
Neeb A, Herranz N, Arce-Gallego S, et al. Advanced prostate cancer with ATM loss: PARP and ATR inhibitors. Eur Urol. 2021;79(2):200–11.
Wu S, Zhou J, Zhang K, et al. Molecular mechanisms of PALB2 function and its role in breast cancer management. Front Oncol. 2020;10:301.
Dillon KM, Bekele RT, Sztupinszki Z, et al. PALB2 or BARD1 loss confers homologous recombination deficiency and PARP inhibitor sensitivity in prostate cancer. NPJ Precis Oncol. 2022;6(1):49.
Carreira S, Porta N, Arce-Gallego S, et al. Biomarkers associating with PARP inhibitor benefit in prostate cancer in the TOPARP-B trial. Cancer Discov. 2021;11(11):2812–27.
van Wilpe S, Simnica D, Slootbeek P, et al. Homologous recombination repair deficient prostate cancer represents an immunologically distinct subtype. Oncoimmunology. 2022;11(1):2094133.
Kamel D, Gray C, Walia JS, Kumar V. PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: an update of clinical trials. Curr Drug Targets. 2018;19(1):21–37.
O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–60.
Morales J, Li L, Fattah FJ, et al. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr. 2014;24(1):15–28.
Davar D, Beumer JH, Hamieh L, Tawbi H. Role of PARP inhibitors in cancer biology and therapy. Curr Med Chem. 2012;19(23):3907–21.
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Sagaram S, Rao A. Rapidly evolving treatment paradigm and considerations for sequencing therapies in metastatic prostate cancer-a narrative review. Transl Androl Urol. 2021;10(7):3188–98.
Risdon EN, Chau CH, Price DK, Sartor O, Figg WD. PARP inhibitors and prostate cancer: to infinity and beyond BRCA. Oncologist. 2021;26(1):e115–29.
EMA. Summary of Product Characteristics. 24 March 2023. https://www.ema.europa.eu/documents/product-information/lynparza-epar-product-information_en.pdf. Accessed 2023 Apr 2023.
Hussain M, Mateo J, Fizazi K, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. 2020;383(24):2345–57.
Abida W, Campbell D, Patnaik A, et al. Rucaparib for the treatment of metastatic castration-resistant prostate cancer associated with a DNA damage repair gene alteration: final results from the phase 2 TRITON2 study. Eur Urol. 2023;84(3):321–30.
Matsubara N, de Bono J, Olmos D, et al. Olaparib efficacy in patients with metastatic castration-resistant prostate cancer and BRCA1, BRCA2, or ATM alterations identified by testing circulating tumor DNA. Clin Cancer Res. 2023;29(1):92–9.
de Bono JS, Mehra N, Scagliotti GV, et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol. 2021;22(9):1250–64.
Reichert ZR, Devitt ME, Alumkal JJ, et al. Targeting resistant prostate cancer, with or without DNA repair defects, using the combination of ceralasertib (ATR inhibitor) and olaparib (the TRAP trial). J Clin Oncol. 2022;40(6_suppl):88–88.
Reichert ZR, Daignault S, Teply BA, Devitt ME, Heath EI. Targeting resistant prostate cancer with ATR and PARP inhibition (TRAP trial): a phase II study. J Clin Oncol. 2020;38(6_suppl):TPS254–TPS254.
Ryan CJ, Abida W, Bryce AH, et al. TRITON3: an international, randomized, open-label, phase III study of the PARP inhibitor rucaparib vs. physician’s choice of therapy for patients with metastatic castration-resistant prostate cancer (mCRPC) associated with homologous recombination deficiency (HRD). J Clin Oncol. 2018;36(6):TPS389–TPS389.
Fizazi K, Piulats JM, Reaume MN, et al. Rucaparib or physician’s choice in metastatic prostate cancer. N Engl J Med. 2023;388(8):719–32.
Agarwal N, Zhang T, Efstathiou E, et al. The biology behind combining poly [ADP ribose] polymerase and androgen receptor inhibition for metastatic castration-resistant prostate cancer. Eur J Cancer. 2023;192:113249.
Clarke N, Wiechno P, Alekseev B, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19(7):975–86.
Agarwal N, Azad A, Carles J, et al. TALAPRO-2: phase 3 study of talazoparib (TALA) + enzalutamide (ENZA) versus placebo (PBO) + ENZA as first-line (1L) treatment in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2023;41:LBA17.
Chi KN, Rathkopf DE, Smith MR, et al. Phase 3 MAGNITUDE study: first results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J Clin Oncol. 2022;40(6_suppl):12–12.
Saad F, Thiery-Vuillemin A, Wiechno P, et al. Patient-reported outcomes with olaparib plus abiraterone versus placebo plus abiraterone for metastatic castration-resistant prostate cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2022;23(10):1297–307.
Clarke NW, Armstrong AJ, Thiery-Vuillemin A, et al. Final overall survival (OS) in PROpel: abiraterone (abi) and olaparib (ola) versus abiraterone and placebo (pbo) as first-line (1L) therapy for metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2023;41:LBA16.
FDA. FDA approves olaparib with abiraterone and prednisone (or prednisolone) for BRCA-mutated metastatic castration-resistant prostate cancer. 2023. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-abiraterone-and-prednisone-or-prednisolone-brca-mutated-metastatic-castration. Accessed 2023 Oct 19.
FDA. FDA approves talazoparib with enzalutamide for HRR gene-mutated metastatic castration-resistant prostate cancer. 2023. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-talazoparib-enzalutamide-hrr-gene-mutated-metastatic-castration-resistant-prostate. Accessed 2023 Oct 19.
Chi KN, Rathkopf D, Attard G, et al. 897TiP - a phase III randomized, placebo-controlled, double-blind study of niraparib plus abiraterone acetate and prednisone versus abiraterone acetate and prednisone in patients with metastatic prostate cancer (NCT03748641). Ann Oncol. 2019;30:v354.
Chi KN, Rathkopf D, Smith MR, et al. Niraparib and abiraterone acetate for metastatic castration-resistant prostate cancer. J Clin Oncol. 2023;41(18):3339–51.
Chi KN, Sandhu S, Smith MR, et al. Niraparib plus abiraterone acetate with prednisone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: second interim analysis of the randomized phase III MAGNITUDE trial. Ann Oncol. 2023;34(9):P772–782. https://doi.org/10.1016/j.annonc.2023.06.009
Rao A, Ryan CJ, VanderWeele DJ, et al. CASPAR (Alliance A031902): a randomized, phase III trial of enzalutamide (ENZ) with rucaparib (RUCA)/placebo (PBO) as a novel therapy in first-line metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2021;39(6_suppl):TPS181–TPS181.
Rathkopf DE, Chi KN, Olmos D, et al. AMPLITUDE: a study of niraparib in combination with abiraterone acetate plus prednisone (AAP) versus AAP for the treatment of patients with deleterious germline or somatic homologous recombination repair (HRR) gene-altered metastatic castration-sensitive prostate cancer (mCSPC). J Clin Oncol. 2021;39(6_suppl):TPS176–TPS176.
Agarwal N, Azad A, Fizazi K, et al. Talapro-3: a phase 3, double-blind, randomized study of enzalutamide (ENZA) plus talazoparib (TALA) versus placebo plus enza in patients with DDR gene mutated metastatic castration-sensitive prostate cancer (mCSPC). J Clin Oncol. 2022;40(6_suppl):TPS221–TPS221.
Karzai F, VanderWeele D, Madan RA, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6(1):141.
Yu EY, Piulats JM, Gravis G, et al. Pembrolizumab plus olaparib in patients with metastatic castration-resistant prostate cancer: long-term results from the phase 1b/2 KEYNOTE-365 cohort a study. Eur Urol. 2023;83(1):15–26.
Yu EY, Kolinsky MP, Berry WR, et al. Pembrolizumab plus docetaxel and prednisone in patients with metastatic castration-resistant prostate cancer: long-term results from the phase 1b/2 KEYNOTE-365 cohort B study. Eur Urol. 2022;82(1):22–30.
Yu EY, Park SH, Goh JCH, et al. 1362MO pembrolizumab + olaparib vs abiraterone (abi) or enzalutamide (enza) for patients (pts) with previously treated metastatic castration-resistant prostate cancer (mCRPC): randomized open-label phase III KEYLYNK-010 study. Ann Oncol. 2022;33:S1163–4.
Markowski MC, Sternberg CN, Wang H, et al. TRIUMPH: phase II trial of rucaparib monotherapy in patients with metastatic hormone-sensitive prostate cancer harboring germline DNA repair gene mutations. J Clin Oncol. 2023;41(6_suppl):190.
Lee A. Fuzuloparib: first approval. Drugs. 2021;81(10):1221–6.
Hussain M, Daignault-Newton S, Twardowski PW, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2018;36(10):991–9.
Agarwal N, Azad AA, Carles J, et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): a randomised, placebo-controlled, phase 3 trial. Lancet. 2023;402(10398):291–303.
Matsubara N, Fizazi K, Azad AA, et al. Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: subgroup analyses of the all-comers cohort from TALAPRO-2 by homologous recombination repair status, in ESMO. Madrid: ESMO; 2023.
Eleni Efstathiou MRS, Sandhu S, Attard G, et al. Niraparib (NIRA) with abiraterone acetate and prednisone (AAP) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and homologous recombination repair (HRR) gene alterations: second interim analysis (IA2) of MAGNITUDE. J Clin Oncol. 2023;41(6_suppl):170.
Antonarakis ES, Park SH, Goh JC, et al. Pembrolizumab plus olaparib for patients with previously treated and biomarker-unselected metastatic castration-resistant prostate cancer: the randomized, open-label, phase III KEYLYNK-010 Trial. J Clin Oncol. 2023;41(22):3839–50.
Kasparian S, Burnham L, Kittles R, et al. Identifying androgen receptor (AR) and genomic characteristics that define populations of patients with mHSPC who benefit from early PARP inhibition therapy with talazoparib in ASCO. J Clin Oncol. 2021. https://doi.org/10.1200/JCO.2021.39.15_suppl.TPS5097.
Zhuang J, Qiu X, Zhang S, Guo H. PROact: a prospective phase II study to investigate the efficacy and safety of olaparib plus abiraterone and prednisone combination therapy in mHSPC patients with HRR gene mutation, in ESMO Asia. Ann Oncol. 2022;33:S1495–502.
Medical Writing/Editorial Assistance
Editorial assistance for this review article was provided by Christos Evangelou PhD and Jake Burrell PhD (Rude Health Consulting Limited), which was funded by MSD China.
Funding
This review article was funded by MSD China and the journal’s Rapid Service Fee was paid for by MSD China.
Author information
Authors and Affiliations
Contributions
All authors substantially contributed to planning, gathering, and interpreting the information or ideas used in the article, including draft revision or critical review, and ensured that questions related to the accuracy or integrity of any part of the work were appropriately resolved. All authors approved the final version of the article.
Corresponding author
Ethics declarations
Conflict of Interest
Zhenhua Liu is employed by MSD China. The other authors report no conflicts of interest.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fan, Y., Liu, Z., Chen, Y. et al. Homologous Recombination Repair Gene Mutations in Prostate Cancer: Prevalence and Clinical Value. Adv Ther 41, 2196–2216 (2024). https://doi.org/10.1007/s12325-024-02844-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-024-02844-7