
Abstract
Introduction
Diroximel fumarate (DRF) is an oral fumarate for relapsing multiple sclerosis (MS) with the same active metabolite as dimethyl fumarate (DMF). DRF has a safety/efficacy profile similar to DMF but with improved gastrointestinal (GI) tolerability and low (< 1%) treatment discontinuation due to GI adverse events (AEs). Efficacy and safety outcomes in patients who switched to DRF from other disease-modifying therapies (DMTs) have not been evaluated.
Methods
EVOLVE-MS-1 is an ongoing, 2-year, open-label, phase 3 study of DRF in adults with relapsing–remitting MS. Patients either entered as newly enrolled to DRF trials, or from the 5-week, randomized, head-to-head, phase 3 EVOLVE-MS-2 study of DRF and DMF. This analysis evaluated safety and GI tolerability in patients continuing on DRF (DRF-rollover) or switching from DMF (DMF-rollover) following EVOLVE-MS-2. Safety and efficacy were evaluated in a subset of newly enrolled patients who had received prior glatiramer acetate (GA; GA/DRF) or interferons (IFN; IFN/DRF) as their most recent DMT, prior to switching to DRF in EVOLVE-MS-1.
Results
As of September 1, 2020, 1057 patients were enrolled in EVOLVE-MS-1, including 166, 182, 239, and 225 patients in the GA/DRF, IFN/DRF, DRF-rollover, and DMF-rollover groups, respectively. Treatment discontinuation due to GI AEs was < 1% in all groups. GA/DRF and IFN/DRF patients experienced improvements from baseline in clinical and radiological efficacy outcomes, including significantly reduced annualized relapse rates. Rollover patients had low rates of new or recurrent GI AEs (DRF-rollover, 26.8%/4.2%; DMF-rollover, 27.1%/4.9%).
Conclusion
After 2 years of DRF exposure, patients with prior GA, IFN, or fumarate treatment had safety outcomes consistent with previous fumarate studies. Efficacy in patients with prior GA or IFN treatment was consistent with previous fumarate studies. The data suggest that transition to DRF from GA, IFN, or DMF is a reasonable treatment strategy, with low rates of discontinuation due to GI AEs.
Trial Registration
ClinicalTrials.gov (NCT02634307).
Infographic

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Diroximel fumarate (DRF) previously demonstrated improved gastrointestinal (GI) tolerability compared with dimethyl fumarate (DMF) in the phase 3, randomized, head-to-head double-blind EVOLVE-MS-2 trial of patients with relapsing–remitting multiple sclerosis (RRMS). |
An interim analysis of the ongoing, open-label, 96-week, phase 3 EVOLVE-MS-1 study of adults with RRMS indicated a low incidence of GI adverse events (AEs) and a very low rate of treatment discontinuation due to GI AEs in the overall study population, but an analysis of DRF outcomes following prior disease-modifying therapy (DMT) has not been conducted. |
The objectives of this analysis were to (1) assess safety and efficacy outcomes in patients with RRMS and up to 2 years of DRF treatment who had received prior glatiramer acetate (GA) or interferon (IFN) as their most recent DMT before initiating DRF in EVOLVE-MS-1, and (2) evaluate GI tolerability in patients who completed 5 weeks of DRF or DMF treatment in EVOLVE-MS-2 before initiating DRF in EVOLVE-MS-1. |
What was learned from the study? |
Safety outcomes in patients who switch to DRF treatment were consistent with what has been reported for the overall population: AEs were generally mild or moderate in severity and led to treatment discontinuation at an overall similar rate among all analysis groups; absolute lymphocyte count decreased by approx. 30% over the first year of treatment with DRF and then stabilized to a level greater than the lower limit of normal for the majority of patients consistent with other DMF studies. |
Patients treated with DRF over 2 years experienced improvements on clinical and radiological efficacy relative to those reported for the preceding treatment period with IFN and GA. Adjusted annualized relapse rate was significantly reduced (p < 0.0001 for both prior IFN and prior GA groups) after up to 2 years of DRF treatment compared with the reported annualized relapse rate in the 12 months before fumarate initiation. |
Patients with prior DMF or DRF treatment for 5 weeks in EVOLVE-MS-2 had low rates of new or recurring GI AEs after initiating DRF in the 2-year EVOLVE-MS-1 study, and the rate of treatment discontinuation due to GI AEs across both groups (prior DMF or prior DRF) was < 1%. The data suggest that transition from DMF to DRF, which have the same active metabolite, is a reasonable treatment strategy for appropriate patients with RRMS. |
Digital Features
This article is published with digital features, including an infographic, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.19086380.
Introduction
Over 20 disease-modifying therapies (DMTs) are currently available for the treatment of relapsing multiple sclerosis (MS), including injectable, infused, and oral medications [1]. Glatiramer acetate (GA) and interferon-beta (IFNβ)-1a/b (including IFNβ1a subcutaneous [SC], IFNβ1a intramuscular [IM], peginterferon-β1a [SC or IM], and IFNβ1b [SC]) are injectable DMTs that serve as common first-line treatment options for patients with MS, in addition to oral therapies such as dimethyl fumarate (DMF) and diroximel fumarate (DRF) [2,3,4,5,6,7]. Injectable therapies have been shown to reduce relapse rates and number of new magnetic resonance imaging (MRI) lesions in patients with MS [3,4,5,6]. Although no direct head-to-head comparisons have been performed between IFN, GA, and DMF, comparative effectiveness studies utilizing propensity score methodology indicate that the effectiveness of injectable therapies is slightly lower than that of DMF, as shown by reduced annualized relapse rate (ARR) with DMF treatment compared with injectable treatments [8, 9]. Some patients treated with an injectable therapy discontinue or switch therapies because of tolerability issues such as injection site reactions and flu-like symptoms, lack of efficacy, safety concerns, or overall treatment burden [2, 10,11,12]. Some patients taking DMF experience gastrointestinal (GI) adverse events (AEs) initially, and sometimes throughout treatment, that may lead to dose interruption or discontinuation [13, 14].
DRF is a next-generation oral fumarate approved for the treatment of patients with relapsing forms of MS [15]. Oral administration of DRF leads to rapid conversion to monomethyl fumarate (MMF), the same pharmacologically active metabolite as DMF. It has been demonstrated that 462 mg of DRF results in MMF systemic exposure that is bioequivalent to 240 mg DMF (Supplementary Material) [16]. Therefore, DRF and DMF have similar efficacy and safety profiles. DMF has demonstrated a favorable benefit–risk profile in clinical and real-world studies of patients with MS [17,18,19,20,21]. As of June 30, 2021, more than 537,000 patients have received DMF, representing more than 1,100,000 patient years of exposure. DRF demonstrated improved GI tolerability compared with DMF in the phase 3, randomized, head-to-head, double-blind, 5-week EVOLVE-MS-2 trial [22]. Prior studies of DMF have indicated that the incidence of GI events is highest in the first month of treatment [23], so the 5-week treatment period was a suitable time period for assessing these outcomes. Patients who completed the 5-week treatment period in EVOLVE-MS-2 were eligible to roll over into EVOLVE-MS-1, an ongoing single-arm, open-label study to evaluate safety, tolerability, and efficacy of DRF over 96 weeks.
Evaluation of DRF safety and efficacy in patients who transition from GA or IFN, and DRF GI tolerability in patients who transition from DMF, may help inform clinical practice and sequencing options. In this analysis, the safety and efficacy of DRF in patients who switched to DRF from other DMTs, specifically IFN or GA in the EVOLVE-MS-1 study, was assessed. GI tolerability was also evaluated for patients treated with DRF in the EVOLVE-MS-1 study, which was compared with GI tolerability when they were treated with DMF in the 5-week EVOLVE-MS-2 study.
The objectives of this analysis were to (1) assess safety, efficacy, and tolerability outcomes in patients with up to 2 years of DRF treatment who had received prior GA or IFN as their most recent DMT before initiating DRF in EVOLVE-MS-1, and (2) evaluate GI tolerability in patients who completed 5 weeks of DMF to DRF treatment in the EVOLVE-MS-2 study before rolling over to EVOLVE-MS-1.
Methods
Study Design and Patients
EVOLVE-MS-1 (NCT02634307) is an ongoing, open-label, phase 3 study assessing safety, tolerability, and efficacy of DRF over 96 weeks in adults with relapsing–remitting MS (RRMS). The study design, DRF dosing regimen, patient population, and study endpoints have been previously described [16]. In brief, patients entered the study either as newly enrolled to the DRF clinical development program or after completing EVOLVE-MS-2 (NCT03093324), a randomized, double-blind, phase 3 study in which patients received DRF or DMF over 5 weeks. Rollover patients from EVOLVE-MS-2 either continued on DRF (DRF-rollover) or switched from DMF to DRF (DMF-rollover).
DRF was administered orally as 462 mg twice daily. For patients newly enrolled to the DRF studies, the dose was titrated as 231 mg twice daily for the first study week, followed by 462 mg twice daily thereafter.
Eligible patients were 18–65 years of age with a confirmed diagnosis of RRMS [24], had no history of clinically significant recurring or active GI symptoms within 3 months of screening, and were neurologically stable with no evidence of relapse within 30 days before screening. Rollover patients had already met the EVOLVE-MS-2 study criteria and rolled over into EVOLVE-MS-1. Complete eligibility criteria have been previously described [16].
The study protocol was reviewed and approved by the Copernicus Group institutional review board and the institutional review board/independent ethics committee for each study site (Supplementary Material) before enrolling patients and releasing drug to investigators. The study was conducted in accordance with the protocol, the International Council for Harmonization Guideline E6, all applicable local regulatory requirements, and ethical principles based on the Declaration of Helsinki. All patients provided their written informed consent before participating in the study.
This study evaluated safety and efficacy in a subgroup of patients from EVOLVE-MS-1 who had previously received GA or IFN as their most recent DMT (GA/DRF and IFN/DRF groups, respectively) before switching to DRF when enrolling in EVOLVE-MS-1. The timing of discontinuing prior GA or IFN before initiating DRF was at the patient’s and investigator’s discretion; no defined washout period was included. The subgroup analysis for GI tolerability was performed in rollover patients, defined as those who had completed 5 weeks of DRF or DMF treatment during the EVOLVE-MS-2 study (DRF-rollover and DMF-rollover groups, respectively) before starting EVOLVE-MS-1. The EVOLVE-MS-1 treatment period began at the time of or within 7 days of the EVOLVE-MS-2 end of treatment visit.
Endpoints Included in the Analysis
Safety and tolerability were assessed in all patients included in the analysis. Assessments included number and percentage of treatment-emergent AEs, GI AEs, serious adverse events (SAEs), and AEs leading to treatment discontinuation. AEs were monitored by the investigator at each study visit and coded using the Medical Dictionary for Regulatory Activities (MedDRA) system organ class and preferred term. GI AEs were defined according to the MedDRA system organ class for GI disorders. A complete blood count, including absolute lymphocyte count (ALC), was assessed at each study visit. ALCs were graded according to the Common Terminology Criteria for Adverse Events (CTCAE v4.0) as follows: grade 0 (≥ lower limit of normal [LLN] [≥ 910 mm3]); grade 1 (< LLN to ≥ 800 mm3); grade 2 (< 800–500 mm3); grade 3 (< 500–200 mm3); and grade 4 (< 200 mm3); ALC LLN was defined as < 0.91 × 109/L [16, 25, 26].
Efficacy outcomes were assessed in the GA/DRF and IFN/DRF groups relative to the outcomes reported for the prior treatment on GA and IFN, including ARR, proportion of patients free from relapse, proportion free from confirmed disability progression (CDP), proportion with confirmed disability improvement (CDI), proportion with no evidence of disease activity (NEDA)-3, patient-reported outcomes (PROs), and MRI outcomes—number of gadolinium-enhancing (Gd+) lesions, percentage of patients free from Gd+ lesions, percentage brain volume change (PBVC), and number of new or enlarging T2 lesions.
On-study relapse endpoints were based on the number of protocol-defined relapses, defined as new or recurrent neurologic symptoms, not associated with fever or infection, lasting for at least 24 h and accompanied by new neurological findings reflected in a change in Expanded Disability Status Scale (EDSS) score. Pre-study relapse endpoints were based on patient-reported number of relapses in the 12 months before study enrollment, collected at baseline. CDP was based on EDSS score and defined as a ≥ 1.5-point increase from a baseline score of 0.0, a ≥ 1.0-point increase from a baseline score of 1.0 to < 6.0, or a ≥ 0.5-point increase from a baseline score of ≥ 6.0, confirmed 12 weeks later. CDI was defined as a ≥ 1.0-point decrease in EDSS score from a baseline EDSS score ≥ 2.0, confirmed 12 weeks later. CDI was not assessed in patients with a baseline EDSS score of < 2.0. NEDA-3 was defined as no relapses, no CDP sustained for 12 weeks as measured on EDSS, no new or enlarging T2 hyperintense lesions, and no new Gd+ lesions, during the 96-week trial duration. Brain MRI scans were performed with and without gadolinium utilizing a standardized sequence under the supervision by the MRI reading center (NeuroRx Research, Montreal, QC, Canada). MRIs were obtained at baseline, 48 weeks, 96 weeks, or at study termination. MRI scans included the following sequence: PD-, T1, and T2-weighted 2D multislice (pre-contrast), T1-weighted 3D spoiled gradient recalled echo (pre-contrast), 2D T2-weighted FLAIR, and 3D spoiled gradient-recalled echo T1-weighted (postcontrast). For assessment of brain atrophy, PBVC was calculated automatically for week 48 and 96 relative to normalized brain volume at baseline. For the PBVC, the type and length of format was BEST 6.2 and the units were percentages in a SAS Dataset format for visits at week 48, 96, and end of study. PROs were assessed using the 12-item Short-Form health survey (SF-12) and the 5-level EuroQol-5d group health outcome measure (EQ-5D-5L). DRF treatment adherence was assessed in all patients and was reported as a percentage, calculated as the number of capsules taken divided by the number of capsules expected to be taken, multiplied by 100.
A GI tolerability analysis was conducted in DRF- and DMF-rollover patients. Analysis of GI tolerability assessments included onset of new GI AEs, recurrence of prior GI AEs, and GI-related treatment discontinuations.
Statistics
Safety and tolerability assessments were conducted in patients who received at least one dose of DRF and summarized using descriptive statistics. MS relapse rate was assessed in patients with prior GA or IFN treatment who received at least one dose of DRF. Adjusted ARR was based on a Poisson regression model adjusted for study period. Adjusted ARR included protocol-defined relapses that occurred during the EVOLVE-MS-1 treatment period (up to 2 years of DRF). Rates were compared with the reported relapse rate for the 12 months before DRF initiation in EVOLVE-MS-1 for GA/DRF and IFN/DRF groups. CDP and MRI outcomes were assessed in patients who received at least one dose of DRF and completed at least one post-baseline efficacy assessment. The estimated proportion of patients free from relapse and free from CDP was based on the Kaplan–Meier product limit method. The estimated proportion of patients with CDI was based on the Kaplan–Meier product limit method and only included patients with a baseline EDSS ≥ 2.0. The estimated proportion of patients with NEDA-3 was based on the Kaplan–Meier product limit method using relapse, MRI, EDSS data, and based on the number of evaluable patients for NEDA-3.
Gd+ lesion count, new or enlarging T2 lesion count, and PBVC were summarized by time point and change from baseline using descriptive statistics. Changes in SF-12 mental and physical component scale scores and EQ-5D-5L index and visual analog scale (VAS) scores from baseline to year 2 were summarized by prior treatment group.
Results
Patients
A total of 1057 patients were enrolled in EVOLVE-MS-1 as of September 1, 2020. This switch subgroup analysis included 166 patients who had received GA (GA/DRF) and 182 patients who had received IFN (IFN/DRF) as their most recent prior DMT before being treated with DRF in EVOLVE-MS-1. In the GI tolerability subgroup analysis, 239 patients previously treated with DRF in EVOLVE-MS-2 (DRF-rollover) and 225 previously treated with DMF in EVOLVE-MS-2 (DMF-rollover) before being treated with DRF in EVOLVE-MS-1 were included.
As of September 2020, 87% (916/1057) of patients completed the 2-year study. Of the patients in the GA/DRF and IFN/DRF groups, 57% (94/166) and 59% (108/182) completed the 2-year study, respectively, and 33% (55/166) and 30% (54/182) discontinued treatment at the time of the analysis, respectively (Fig. 1). Of the patients in the DRF-rollover and DMF-rollover groups, 40.2% (96/239) and 42.2% (95/225) had completed the study, respectively, and 26.4% (63/239) and 25.8% (58/225) had discontinued treatment, respectively.

Patient disposition. aIncludes pregnancy, physician decision, lack of efficacy, noncompliance with study drug, or other reason. Of the patients enrolled in EVOLVE-MS-1, only those who were previously treated with IFN or GA, or who rolled over from the completed EVOLVE-MS-2 study were included in this analysis. AE adverse event, DMF dimethyl fumarate, DRF diroximel fumarate, GA glatiramer acetate, IFN interferon
Baseline characteristics were generally similar among all analysis groups, although patients switching from IFN had a slightly longer time since diagnosis than those from the other analysis groups (Table 1). Patients in the GA/DRF and IFN/DRF groups received prior GA or IFN for a median (interquartile range [IQR]) treatment duration of 1.76 (0.82–3.55) and 2.80 (0.86–5.58) years, respectively, before initiating DRF in EVOLVE-MS-1. Patients in the DRF-rollover and DMF-rollover groups had received DRF or DMF for 5 weeks as mandated by the EVOLVE-MS-2 protocol before initiating DRF in EVOLVE-MS-1.
At the time of this analysis, median (range) duration of DRF treatment was 1.8 (0.0–1.9) years for the GA/DRF and IFN/DRF groups and 1.6 (0.0–1.9) years for the DRF-rollover and DMF-rollover groups. Mean (standard deviation [SD]) treatment adherence with DRF was above 93% for all groups: GA/DRF 93.1% (11.6), IFN/DRF 94.5% (9.9), DRF-rollover 95.0% (9.1), and DMF-rollover 95.4% (7.7).
Safety
Safety Summary
The percentage of patients with any treatment-emergent AE was similar across all analysis groups: GA/DRF, 92.8%; IFN/DRF, 88.5% DRF-rollover, 87.9%; DMF-rollover, 90.2% (Table 2). AEs were mild or moderate in severity for most patients. The most common AEs in patients with GA/DRF or IFN/DRF treatment were flushing (32.5% and 33.0%), MS relapse (23.5% and 15.9%), upper respiratory tract infection (15.1% and 16.5%), and diarrhea (15.1% and 11.0%), respectively (Table 3). The most common AEs in patients with prior fumarate exposure in the DRF-rollover and DMF-rollover groups, respectively, were MS relapse (19.2% and 18.7%), upper respiratory tract infection (16.7% and 15.6%), lymphopenia (13.8% and 15.1%), and flushing (13.8% and 12.9%). Rollover patients had a lower incidence of GI AEs (DRF-rollover, 29.3% [70/239]; DMF-rollover, 28.4% [64/225]) compared with patients whose most recent DMT was not a fumarate (GA/DRF, 34.9%; IFN/DRF, 37.9%).
Infection was reported by approximately 50% of patients, including 50.0%, 48.9%, 45.2%, and 52.9% of GA/DRF, IFN/DRF, DRF-rollover, and DMF-rollover patients, respectively (Table 2). Rates of serious infections, opportunistic infections (Candida infections), and confirmed/suspected COVID-19 were at most 1% across all patient groups.
AEs led to discontinuation of study drug in 11.4% of GA/DRF and 10.4% of IFN/DRF patients. Rates were slightly lower in rollover patients: 9.6% DRF-rollover, 5.8% DMF-rollover. GI AEs resulted in treatment discontinuation in three of the 811 patients (< 1%) across all groups: IFN/DRF, n = 1; DRF-rollover, n = 1; DMF-rollover, n = 1. Flushing resulted in treatment discontinuation in six of the 811 patients (< 1%) across all groups: GA/DRF, n = 2; IFN/DRF, n = 1; DRF-rollover, n = 2; DMF-rollover, n = 1.
Change in Absolute Lymphocyte Count
Across all analysis groups, ALC declined by approximately 30% over the first year of treatment (GA/DRF, 31.7%; IFN/DRF, 27.1%; DRF-rollover, 29.0%; DMF-rollover, 30.2%) and then stabilized to a level above the LLN (Fig. 2) for the majority of patients in all groups: GA/DRF 57.0% (94/165), IFN/DRF 52.8% (93/176), DRF-rollover 58.8% (140/238), DMF-rollover 60.4% (136/225). Approximately 10% of patients experienced at least one ALC < 0.5 × 109/L: 10.3% (17/165) in the GA/DRF; 12.5% (22/176) in the IFN/DRF; 10.5% (25/238) in the DRF-rollover; and 9.3% (21/225) in the DMF-rollover groups. The protocol stop** rule was patients who had a lymphocyte count < 0.5 × 109/L had to temporarily discontinue and if the ALC remained < 0.5 × 109/L then the subject must permanently discontinue; therefore, the number of patients with severe prolonged (> 6 months) lymphopenia was not assessed.

Change in ALC over time. aPatients in the DRF-rollover and DMF-rollover groups received 5 weeks of DRF or DMF treatment before EVOLVE-MS-1 enrollment. ALC absolute lymphocyte count, DMF dimethyl fumarate, DRF diroximel fumarate, LLN lower limit of normal
Efficacy
Clinical Efficacy in Patients Initiating DRF After Prior GA or IFN Treatment
Adjusted ARR was significantly reduced (GA/DRF 74.0% [rate ratio 0.26, 95% confidence interval 0.18, 0.38] and IFN/DRF 78.7% [rate ratio 0.21, 95% confidence interval 0.14, 0.31]; p < 0.0001 for both) during the EVOLVE-MS-1 treatment period (DRF for up to 2 years) compared with the 12 months before treatment with DRF (Fig. 3). The estimated proportion of patients in the GA/DRF and IFN/DRF groups who were free from relapse at week 96 was 78.4% and 85.5%, respectively. The estimated proportion of patients who were free from CDP at week 96 was 84.9% and 87.4%, respectively. The estimated proportion of patients with CDI at week 96 was 16.3% and 10.3%, respectively. The estimated proportion of patients with NEDA-3 at week 48 and week 96 (over the full 96-week treatment period) was GA/DRF, 63.0% and 39.9%; and IFN/DRF, 69.9% and 45.8%, respectively (Fig. 4).

Adjusted ARR on DRF treatment compared with the 12 months before DRF or DMF initiation in GA/DRF and IFN/DRF treatment groups: adjusted ARR for protocol-defined relapses during the EVOLVE-MS-1 treatment period (up to 2 years of DRF treatment) compared with patient-reported relapses in the 12 months before DRF initiation in EVOLVE-MS-1. ARR annualized relapse rate, DRF diroximel fumarate, GA glatiramer acetate, IFN interferon

Estimated proportion of patients with NEDA-3 after 1 or 2 years of DRF treatment in EVOLVE-MS-1. DRF diroximel fumarate, GA glatiramer acetate, IFN interferon, NEDA-3 no evidence of disease activity-3
Radiological Efficacy in Patients Initiating DRF After Prior GA or IFN Treatment
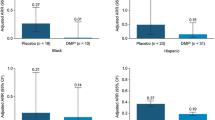
For all analysis groups, mean number of Gd+ lesions decreased over the 2-year treatment period relative to prior GA or IFN treatment (GA/DRF and IFN/DRF groups) (Fig. 5a). The percentage of patients free from Gd+ lesions increased from the relative baseline to year 2 in the GA/DRF (from 78.8% to 96.0%) and IFN/DRF groups (from 80.2% to 92.8%; Fig. 5b). Mean (SD) number of new or enlarging T2 lesions from the relative baseline to year 1 and from year 1 to year 2 was 1.0 (2.5) and 0.3 (1.4), respectively, in the GA/DRF; and 1.7 (6.6) and 1.3 (6.0), respectively, in the IFN/DRF groups. Mean (SD) PBVC from year 1 to year 2 was − 0.2 (0.6) in the GA/DRF group; and − 0.3 (0.6) in the IFN/DRF group (Fig. 6).

a Mean (SE) number of Gd+ lesions and b percentage of patients free from Gd+ lesions. DRF diroximel fumarate, GA glatiramer acetate, Gd+ gadolinium-enhancing, IFN interferon

Mean (SE) PBVC from year 1 to year 2a in EVOLVE-MS-1. aPBVC from baseline to year 1 is not reported to avoid confounding by pseudo-atrophy. bEstimated yearly brain volume loss in healthy adults [35]. DRF diroximel fumarate, GA glatiramer acetate, IFN interferon, PBVC percentage brain volume change
Patient-Reported Outcomes
SF-12 mental and physical component scores and EQ-5D index and VAS scores remained stable over the 2-year treatment period for both GA/DRF and IFN/DRF groups (Table 4).
GI Tolerability in EVOLVE-MS-2 Rollover Patients
A total of 33.9% (81/239) and 44.9% (101/225) of patients in the DRF-rollover and DMF-rollover groups, respectively, had a prior GI AE in EVOLVE-MS-2 (Table 5). Most of these AEs were mild or moderate in severity (96.3% [78/81] and 97.0% [98/101], respectively). The events had resolved in 77.8% (63/81) and 78.2% (79/101) of patients, respectively; GI AEs that resolved in EVOLVE-MS-2 were unlikely to reoccur in the EVOLVE-MS-1 extension study (DRF-rollover, 4.2%; DMF-rollover, 4.9%). Onset of new GI AEs (the report of a GI AE not previously reported in the EVOLVE-MS-2 study) in EVOLVE-MS-1 was 26.8% in the DRF-rollover group and 27.1% in the DMF-rollover group. Most were mild or moderate in severity (93.8% [60/64] and 91.8% [56/61], respectively).
Overall, the rates of GI AEs in EVOLVE-MS-1 were 29.3% (70/239) and 28.4% (64/225) for DRF-rollover and DMF-rollover patients, respectively. The majority of events were mild or moderate in severity: DRF-rollover, 94.3% (66/70); DMF-rollover 90.6% (58/64). Patients in the DMF-rollover group had a slightly higher incidence of diarrhea (DMF-rollover 11.1% vs DRF-rollover 7.5%), nausea (DMF-rollover 6.7% vs DRF-rollover 5.0%), constipation (DMF-rollover 5.3% vs DRF-rollover 3.8%), and vomiting (DMF-rollover 4.4% vs DRF-rollover 2.9%) compared with patients in the DRF-rollover group. Fewer than 1% of all patients with GI AEs in EVOLVE-MS-2 discontinued treatment because of GI AEs in EVOLVE-MS-1 (Table 6).
Discussion
Patients with up to 2 years of DRF treatment who had rolled over from EVOLVE-MS-2 or had received GA or IFN as their most recent DMT before study enrollment had overall safety outcomes that were consistent with previous studies of DRF [16, 22], and there were no unexpected safety concerns. For patients who received GA or IFN as their most recent DMT (GA/DRF and IFN/DRF groups), significant improvements in ARR were observed on DRF for up to 2 years compared with the 12 months before treatment with DRF (p < 0.0001 for both). Improvements in other clinical and radiological measures of disease activity were observed on DRF, but were not found to be statistically significant. Thus, the findings in this study support previous studies demonstrating that oral fumarates may be an effective treatment option for patients switching from GA or IFNs [11, 21, 27].
AEs were generally mild or moderate in severity and led to treatment discontinuation at an overall similar rate among patients in this analysis and the overall study population [16]. GI AEs and flushing both occurred at slightly higher rates in patients with no prior fumarate exposure compared with patients who received 5 weeks of DRF or DMF in EVOLVE-MS-2. This finding is not unexpected given that GI AEs and flushing are known to occur early in treatment, typically within 1 month of DRF or DMF initiation and decrease over time [13, 16]. Therefore, patients with prior fumarate exposure who may have been susceptible to flushing and GI AEs likely experienced the majority of those events during the earlier EVOLVE-MS-2 study.
An approximately 30% decline in ALC was observed over the first year of treatment across all treatment groups and plateaued over time. This change in ALC was also observed in the phase 3 and extension studies for DMF [23], and was previously observed with DRF in the overall population of EVOLVE-MS-1 [16]. Despite the decline in ALC, previous studies have reported an intact humoral immune response in DMF-treated patients with RRMS, as demonstrated by the immune response (recall antigen, T cell-independent antigen, and neoantigen) to vaccination that provided adequate seroprotection [28], and stable levels of immunoglobulins (IgA, IgG, and IgM) over 2 years of therapy regardless of ALCs [29]. These findings are consistent with long-term safety data that show no overall increased risk of serious infections or malignancies with DMF treatment, with the exception of progressive multifocal leukoencephalopathy cases [25]. COVID-19 occurred in less than 1% of patients (one patient with prior IFN treatment and two patients with prior DMF exposure), in line with studies showing fumarate, GA, and IFN DMTs have not been associated with higher risk of COVID-19 complications [30,31,32].
After up to 2 years of DRF treatment, numerous patients in the GA/DRF and IFN/DRF groups experienced improvements from baseline in clinical and radiological efficacy outcomes relative to prior treatment. ARR was significantly lower with up to 2 years of DRF treatment compared with the 12 months before DRF (GA/DRF and IFN/DRF groups) initiation, with similar percentage reductions in both analysis groups and the overall population [16]. The proportion of patients with NEDA-3 was approximately 70% at week 48 and approximately 40% after 96 weeks of therapy in patients with prior GA or IFN treatment, rates that are consistent with the observed 1-year and 2-year rates with DMF and DRF [16, 33, 34]. Comparisons of efficacy in patients on DMF in EVOLVE-MS-2 versus when they were on DRF in EVOLVE-MS-1 could not be conducted as a result of limited exposure in EVOLVE-MS-2 (5 weeks) but the outcomes are likely to be similar for DMF because DRF and DMF administration results in bioequivalent exposures of the same active metabolite, MMF (Supp. Fig.) [16]. However, if a patient wants to switch DMTs due to a non-efficacy reason, DRF may be an appropriate choice because of its improved tolerability while likely maintaining efficacy.
Patients in this analysis had fewer Gd+ lesions over the 2-year study period compared with baseline and a greater proportion were Gd+ lesion-free with DRF treatment (GA/DRF, 96.0%; IFN/DRF, 92.8%), with overall reductions similar to the observed rates in the overall population [16]. In addition, patients treated with DRF over 2 years showed a mean yearly brain volume change that was similar to annualized rates of brain atrophy of − 0.27 ± 0.15% in healthy adults (Fig. 6) [35].
Patient-centered factors are important considerations when selecting a treatment regimen, especially addressing patient’s lifestyle, treatment burden, and interference in daily activities. In this analysis, PROs indicated that quality of life (mobility, self-care, usual activities, pain/discomfort, anxiety/depression) remained stable over the 2-year treatment period for both analysis groups. These findings are consistent with those of the EVOLVE-MS-1 interim analysis [16], health-related quality-of-life data reported with other DMTs [36], and the EVOLVE-MS-2 secondary analysis, which indicated treatment with DRF was associated with positive quality-of-life outcomes, including reduced effect on daily life and work and less concomitant symptomatic medication use (e.g., proton pump inhibitors, anticholinergics/antispasmodics) [37].
Data from our GI tolerability analysis of EVOLVE-MS-2 rollover patients demonstrate that patients who switched from DMF to DRF (DMF-rollover group) had a low incidence of new or recurrent GI AEs with DRF over the 2-year treatment period, with rates similar to those observed in patients treated continuously with DRF (DRF-rollover group). However, the most common AEs, diarrhea and nausea, were reported with higher frequency in the DMF-rollover group than in the DRF-rollover group. Importantly, the rate of treatment discontinuation due to GI AEs was extremely low (< 1%) in EVOLVE-MS-2 rollover patients, regardless of prior fumarate treatment, and was consistent with previous studies of DRF [16, 22, 38]. For reference, the overall rate of treatment discontinuation due to GI AEs was 0.8% (5/593) in all fumarate-naïve patients [Data on file, Biogen], and in the EVOLVE-MS-2 study was 0.8% and 4.8% with DRF or DMF, respectively [22]. Although patients who experienced GI-related treatment discontinuation in EVOLVE-MS-2 would not have rolled over to EVOLVE-MS-1, overall the data indicate that transition from DMF to DRF is a reasonable treatment strategy associated with low rates of discontinuation.
The reported analysis is limited in that efficacy and safety outcomes could be less conclusively assessed relative to baseline because of the open-label study design relying on retrospective data collection of events before therapy. Patients switching from IFN had a slightly longer time from MS diagnosis than those who had received other DMTs. In addition, patients received GA, IFN, or DMF treatment for varying durations before EVOLVE-MS-1 enrollment, and therefore, comparisons cannot be made between the prior treatment groups (GA/DRF and IFN/DRF) and the EVOLVE-MS-2 rollover groups (DRF-rollover and DMF-rollover). Specifically, GA/DRF and IFN/DRF patients received GA or IFN for a median of 1.76 and 2.80 years, respectively, before initiating DRF in EVOLVE-MS-1, whereas EVOLVE-MS-2 rollover patients received DRF or DMF for 5 weeks after previously being on no DMT (DRF-rollover, 33.2%; DMF-rollover, 33.9%) versus prior DMT (DRF-rollover, 66.8%; DMF-rollover 66.1%). Patients identified as receiving GA or IFN during screening visit could discontinue prior therapy and start DRF between 1 and 28 days before DRF initiation on the basis of the investigator’s discretion.
Conclusion
In this post hoc analysis, patients with RRMS who received up to 2 years of DRF treatment after switching from GA, IFN, or after completing EVOLVE-MS-2 had overall safety outcomes that were consistent with previous studies of fumarates. Notably, the rate of treatment discontinuation due to GI AEs was low (< 1%) in all analysis groups. Patients who switched from GA or IFN also experienced improvements from baseline in clinical and radiological efficacy outcomes consistent with previous studies of fumarates, with just under half of patients (approx. 40%) with NEDA-3 at 2 years and PBVC approaching rates of healthy adults. Patients switching from DMF to DRF experienced low rates of new or recurring GI AEs. The data suggest that transition to DRF from GA, IFN, or DMF is a reasonable treatment strategy with low rates of discontinuation.
References
Disease-modifying therapies for MS. 2021. https://www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/Brochure-The-MS-Disease-Modifying-Medications.pdf. Accessed May 11, 2021.
Fox RJ, Salter AR, Tyry T, et al. Treatment discontinuation and disease progression with injectable disease-modifying therapies: findings from the North American Research Committee on Multiple Sclerosis database. Int J MS Care. 2013;15(4):194–201.
Copaxone (glatiramer acetate) Prescribing Information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/020622s102lbl.pdf. Accessed Feb 22, 2021.
Rebif (interferon beta-1a) Prescribing Information. 2014. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/103780s5178s5179lbl.pdf. Accessed Feb 22, 2021.
Avonex (interferon beta-1a) Prescribing Information. 2020. https://www.avonex.com/content/dam/commercial/avonex/pat/en_us/pdf/Avonex_US_Prescribing_Information.pdf. Accessed Feb 22, 2021.
Betaseron (interferon beta-1b) Prescribing Information. 2020. https://labeling.bayerhealthcare.com/html/products/pi/Betaseron_PI.pdf. Accessed Feb 22, 2021.
Plegridy (peginterferon beta-1a) Prescribing Information. 2021. https://www.plegridyhcp.com/content/dam/commercial/plegridy/hcp/en_us/pdf/prescribing-information.pdf. Accessed May 11, 2021.
Hutchinson M, Fox RJ, Havrdova E, et al. Efficacy and safety of BG-12 (dimethyl fumarate) and other disease-modifying therapies for the treatment of relapsing-remitting multiple sclerosis: a systematic review and mixed treatment comparison. Curr Med Res Opin. 2014;30(4):613–27.
Boster A, Nicholas J, Wu N, et al. Comparative effectiveness research of disease-modifying therapies for the management of multiple sclerosis: analysis of a large health insurance claims database. Neurol Ther. 2017;6(1):91–102.
Daugherty KK, Butler JS, Mattingly M, Ryan M. Factors leading patients to discontinue multiple sclerosis therapies. J Am Pharm Assoc (2003). 2005;45(3):371–5.
Ontaneda D, Nicholas J, Carraro M, et al. Comparative effectiveness of dimethyl fumarate versus fingolimod and teriflunomide among MS patients switching from first-generation platform therapies in the US. Mult Scler Relat Disord. 2019;27:101–11.
Cohan SL, Hendin BA, Reder AT, et al. Interferons and multiple sclerosis: lessons from 25 years of clinical and real-world experience with intramuscular interferon β-1a (Avonex). CNS Drugs. 2021;35(7):743–67.
Phillips JT, Selmaj K, Gold R, et al. Clinical significance of gastrointestinal and flushing events in patients with multiple sclerosis treated with delayed-release dimethyl fumarate. Int J MS Care. 2015;17(5):236–43.
Phillips JT, Agrella S, Fox RJ. Dimethyl fumarate: a review of efficacy and practical management strategies for common adverse events in patients with multiple sclerosis. Int J MS Care. 2017;19(2):74–83.
Vumerity Prescribing Information. 2020. https://www.vumerityhcp.com/content/dam/commercial/vumerity/hcp/en_us/pdf/vumerity-prescribing-information.pdf. Accessed Feb 22, 2021.
Naismith RT, Wolinsky JS, Wundes A, et al. Diroximel fumarate (DRF) in patients with relapsing-remitting multiple sclerosis: interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study. Mult Scler. 2020;26(13):1729–39.
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–107.
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–97.
Gold R, Arnold DL, Bar-Or A, et al. Safety and efficacy of delayed-release dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: 9 years’ follow-up. Ther Adv Neurol Disord. 2020;13:1756286420915005.
Berger T, Brochet B, Brambilla L, et al. Effectiveness of delayed-release dimethyl fumarate on patient-reported outcomes and clinical measures in patients with relapsing–remitting multiple sclerosis in a real-world clinical setting: PROTEC. Mult Scler J Exp Transl Clin. 2019;5(4):2055217319887191.
Kresa-Reahl K, Repovic P, Robertson D, Okwuokenye M, Meltzer L, Mendoza JP. Effectiveness of delayed-release dimethyl fumarate on clinical and patient-reported outcomes in patients with relapsing multiple sclerosis switching from glatiramer acetate: RESPOND, a prospective observational study. Clin Ther. 2018;40(12):2077–87.
Naismith RT, Wundes A, Ziemssen T, et al. Diroximel fumarate demonstrates an improved gastrointestinal tolerability profile compared with dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. CNS Drugs. 2020;34(2):185–96.
Fox RJ, Chan A, Gold R, et al. Characterizing absolute lymphocyte count profiles in dimethyl fumarate-treated patients with MS: patient management considerations. Neurol Clin Pract. 2016;6(3):220–9.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302.
Mehta D, Miller C, Arnold DL, et al. Effect of dimethyl fumarate on lymphocytes in RRMS: implications for clinical practice. Neurology. 2019;92(15):e1724–38.
National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0. 2009. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. Accessed Nov 3, 2021.
Repovic P, Robertson D, Kresa-Reahl K, et al. Effectiveness of dimethyl fumarate in patients with relapsing multiple sclerosis switching after suboptimal response to glatiramer acetate, including patients with early multiple sclerosis: subgroup analysis of RESPOND. Neurol Ther. 2021;10(1):169–82.
von Hehn C, Howard J, Liu S, et al. Immune response to vaccines is maintained in patients. Neurol Neuroimmunol Neuroinflamm. 2018;5(1): e409.
Longbrake EE, Mao-Draayer Y, Cascione M, et al. Dimethyl fumarate treatment shifts the immune environment toward an anti-inflammatory cell profile while maintaining protective humoral immunity. Mult Scler. 2020. https://doi.org/10.1177/1352458520937282.
Reder AT, Centonze D, Naylor ML, et al. COVID-19 in patients with multiple sclerosis: associations with disease-modifying therapies. CNS Drugs. 2021;35(3):317–30.
Sormani MP, De Rossi N, Schiavetti I, et al. Disease-modifying therapies and coronavirus disease 2019 severity in multiple sclerosis. Ann Neurol. 2021;89(4):780–9.
Salter A, Fox RJ, Newsome SD, et al. Outcomes and risk factors associated with SARS-CoV-2 infection in a North American registry of patients with multiple sclerosis. JAMA Neurol. 2021;78(6):699–708.
Havrdova E, Giovannoni G, Gold R, et al. Effect of delayed-release dimethyl fumarate on no evidence of disease activity in relapsing-remitting multiple sclerosis: integrated analysis of the phase III DEFINE and CONFIRM studies. Eur J Neurol. 2017;24(5):726–33.
Mallucci G, Annovazzi P, Miante S, et al. Two-year real-life efficacy, tolerability and safety of dimethyl fumarate in an Italian multicentre study. J Neurol. 2018;265(8):1850–9.
De Stefano N, Stromillo ML, Giorgio A, et al. Establishing pathological cut-offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(1):93–9.
Jongen PJ. Health-related quality of life in patients with multiple sclerosis: impact of disease-modifying drugs. CNS Drugs. 2017;31(7):585–602.
Wundes A, Wray S, Gold R, et al. Improved gastrointestinal profile with diroximel fumarate is associated with a positive impact on quality of life compared with dimethyl fumarate: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. Ther Adv Neurol Disord. 2021;14:1756286421993999.
Liseno J, Lager B, Miller C, Shankar SL, Mendoza JP, Lewin JB. Multiple sclerosis patients treated with diroximel fumarate in the real-world setting have high rates of persistence and adherence. Neurol Ther. 2021. https://doi.org/10.1007/s40120-021-00242-7.
Acknowledgements
The authors thank all participants for their involvement in the study.
Funding
This study was funded by Biogen (Cambridge, MA, USA). Biogen also provided funding for the journal’s Rapid Service Fee and Open Access Fee.
Medical Writing, Editorial, and Other Assistance
Mikhail Melikov (Cytel) provided support for the statistical analyses (funded by Biogen). Biogen provided funding for medical writing support in the development of this manuscript; Rachel Mason, CMPP and Susan Chow, PhD, CMPP from Excel Scientific Solutions wrote the first draft of the manuscript based on input from authors, and Leonard Wills from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. The authors had full editorial control of the manuscript and provided their final approval of all content.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
Jelena Drulovic, Robert T. Naismith, Hailu Chen, Shivani Kapadia, and Jason P. Mendoza contributed to the study conception and design. Material preparation and data collection were performed by Sibyl Wray, Florian Then Bergh, Annette Wundes, Douglas L. Arnold, Elizbieta Jasinska, Donald Negroski, James D. Bowen, Robert T. Naismith, Samuel F. Hunter, and Barry A. Singer. Analyses and interpretation of data were performed by Sibyl Wray, Florian Then Bergh, Annette Wundes, Douglas L. Arnold, Elzbieta Jasinska, Robert T. Naismith, Samuel F. Hunter, Hailu Chen, Jennifer Lyons, Sai L. Shankar, Shivani Kapadia, Jason P. Mendoza, and Brian A. Singer. The first draft of the manuscript was written by Rachel Mason and Susan Chow, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Prior Presentation
A portion of these data have been presented at the Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS) Forum, Virtual Annual Meeting, February 25–27, 2021: Posters P045 and P028.
Disclosures
Sibyl Wray reports consulting fees from and advisory boards for Biogen, Celgene, and EMD Serono; speaker bureaus for Biogen, Celgene, EMD Serono, Genentech-Roche, and Sanofi-Genzyme; research support from Biogen, Celgene, EMD Serono, Genentech-Roche, Novartis, Receptos, Sanofi-Genzyme, and TG Therapeutics. Florian Then Bergh reports grant support from the German Research Foundation (DFG) and Federal Ministry of Education and Research (BMBF); he has received, through his institution, grant support and travel support to attend scientific meetings from Actelion, Bayer, Biogen, Genzyme, Merck-Serono, and Novartis; speaker fees from and advisory boards for Actelion, Bayer, Genzyme, Merck-Serono, Novartis, and Roche. Annette Wundes reports advisor fees from AbbVie; research support from AbbVie, Alkermes, and Biogen. Douglas L. Arnold reports consulting fees from Albert Charitable Trust, Alexion Pharma, Biogen, Celgene, Frequency Therapeutics, Genentech, Med-Ex Learning, Merck, Novartis, Population Council, Receptos, Roche, and Sanofi-Aventis; grants from Biogen, Immunotec, and Novartis; and an equity interest in NeuroRx. Jelena Drulovic reports speaker fees from and advisory boards for Bayer, Hemofarm, Medis, Merck, Novartis, Roche, Sanofi-Genzyme, Biogen, Zentiva, and Teva. Elzbieta Jasinska reports advisory boards for Biogen; speaker fees from Biogen, Novartis, Roche, and Sanofi. James D. Bowen reports speaker/consulting/advisory fees from Alexion, Alkermes, Biogen, Celgene, EMD Serono, Genentech, Genzyme, Novartis, and TG Therapeutics; holds stock in Amgen; research support from Alexion, Alkermes, Biogen, Celgene, Genzyme, Genentech, Novartis, and TG Therapeutics. Donald Negroski reports research support from and consultant/advisory boards/speaker bureaus for Adamas, Alkermes, Alexion, Bayer, Biogen, Celgene/Bristol Myers Squibb, EMD Serono, Genentech-Roche, Janssen Pharmaceuticals, Novartis, and Sanofi-Genzyme. Robert T. Naismith reports speaker/consulting/advisory fees from Alexion, Alkermes, Bayer, Biogen, Celgene, EMD Serono, Genentech, Lundbeck, NervGen, Novartis, Sanofi-Genzyme, Third Rock Therapeutics, and Viela Bio. Samuel F. Hunter reports consulting fees from AbbVie, Janssen-Actelion, Adamas, Biogen, BMS, Novartis, Osmotica, and Sanofi; research support from AbbVie, Actelion, Anokion, Biogen-Alkermes, BMS-Celgene, Novartis, Roche-Genentech, and Sanofi-Genzyme; speaker bureaus for Biogen, BMS, Roche-Genentech, and Sanofi-Genzyme. Mark Gudesblatt reports consulting fees from Biogen, EMD Serono, Novartis, and Sanofi-Genzyme; research support from Alkermes; speaker bureaus for Biogen, EMD Serono, Genentech-Roche, and Sanofi-Genzyme. Hailu Chen, Jennifer Lyons, Sai L. Shankar, Shivani Kapadia, and Jason P. Mendoza are full-time employees of and hold stock/stock options in Biogen. Barry A. Singer reports speaker/consulting fees from AbbVie, Alexion, Biogen, Bristol Myers Squibb, EMD Serono, Janssen, Genentech, Greenwich Biosciences, Novartis, Roche, Sanofi-Genzyme, and TG Therapeutics; research support from AbbVie, Alkermes, Biogen, Greenwich Biosciences, MedImmune, Novartis, Roche, and Sanofi-Genzyme.
Compliance with Ethics Guidelines
The study protocol was reviewed and approved by the Copernicus Group institutional review board and the institutional review board/independent ethics committee for each site (Supplementary Material) before enrolling subjects and releasing drug to investigators. The study was conducted in accordance with the protocol, the International Council for Harmonization Guideline E6, all applicable local regulatory requirements, and ethical principles based on the Declaration of Helsinki. All patients provided written informed consent before participation in the study.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available because the study is still ongoing.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wray, S., Then Bergh, F., Wundes, A. et al. Efficacy and Safety Outcomes with Diroximel Fumarate After Switching from Prior Therapies or Continuing on DRF: Results from the Phase 3 EVOLVE-MS-1 Study. Adv Ther 39, 1810–1831 (2022). https://doi.org/10.1007/s12325-022-02068-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-022-02068-7