
Abstract
Among the 2-arylbenzofuran derivatives isolated from Morus alba, the farnesylated 2-arylbenzofuran is a rarer constituent. The derivative has been reported to exert anti-obesity effect; however, its inhibitory effect on protein tyrosine phosphatase 1B (PTP1B) has not been investigated. In the previous study, the presence of the farnesyl group in the structure of 2-arylbenzofurans was found to have positive influences on their pancreatic lipase inhibitory activity. In the present study, we have confirmed the authenticity of the notation based on the PTP1B inhibitory activity of farnesylated 2-arylbenzofurans. Specifically, two farnesylated 2-arylbenzofurans [morusalfurans B (2) and C (3)] showed strong inhibitory effects on PTP1B with IC50 values of 8.92 and 7.26 µM, respectively, which was significantly higher than that of the positive controls [sodium orthovanadate (IC50 = 15.10 µM) and ursolic acid (IC50 = 11.34 µM)]. Besides, two 2-arylbenzofurans [morusalfurans A (1) and F (6)], one flavonoid [morusalnol B (9)], and one geranylated stilbene [morusibene A (11)] exhibited PTP1B inhibition with IC50 values ranging from 11.02 to 26.56 µM. Kinetic studies revealed compounds 2, 3, 6, and 11 as mixed type PTP1B inhibitors, while 1 and 9 are known as noncompetitive. Molecular docking simulations demonstrated that these active compounds can bind with the respective catalytic or/and allosteric sites of PTP1B with negative binding energies and the results are in accordance with that of the kinetic studies. To the best of our knowledge, this is the first time, the PTP1B inhibitory activity of eleven compounds (1–11), as well as the mechanism of action underlying the effects on PTP1B enzyme of the active compounds, were investigated. In vitro and in silico results suggest that the farnesylated 2-arylbenzofurans from M. alba may potentially be utilized as an effective treatment therapy for type 2 diabetes mellitus and its associated complications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nowadays, changes in human behavior and lifestyle have led to a dramatic increase in the incidence of diabetes mellitus (DM) worldwide (World Health Organization 2016). Diabetes mellitus is a metabolic disease characterized by high blood sugar levels over a prolonged period, it was reported as one of the most common and severe diseases in the world. According to the World Health Organization (WHO), since 1980, the global prevalence of diabetes has nearly doubled among adults over 18 years of age (from 4.7 in 1980 to 8.5% in 2014). In 2012, diabetes directly caused 1.5 million deaths and high blood glucose was responsible for an additional 2.2 million deaths as a result of increased risks of cardiovascular and other diseases (World Health Organization 2016). There are two primary forms of diabetes. Type 1 diabetes mellitus (insulin-dependent DM) is caused by autoimmune destruction of the insulin-producing ß-cells of the pancreatic islets, usually leading to absolute insulin deficiency (Alberti and Zimmet 1998). Type 2 diabetes mellitus (non-insulin-dependent DM, T2DM) is characterized by insulin resistance and relative insulin deficiency, which accounts for about 90 to 95 % of all diagnosed diabetes cases. It is often, but not always, associated with overweight or obesity, which itself can cause insulin resistance and lead to high blood glucose levels (Alberti and Zimmet 1998; Moller 2001). Currently, used insulin sensitizers still have many side effects including weight gain (Moller 2001). An ideal treatment for T2DM would result in the alleviation of the insulin-resistant state combined with weight loss. A specific drug target offering both of these approaches is protein tyrosine phosphatase 1B (PTP1B).
Protein tyrosine phosphatase 1B is an intracellular protein tyrosine phosphatase expressed in insulin-responsive tissues. PTP1B plays a critical role in the insulin receptor signaling pathway. PTP1B directly interacts with activated insulin receptor or insulin receptor substrate-1 to dephosphorylate phosphotyrosine residues, resulting in the down-regulation of insulin action (Elchebly et al. 1999). PTP1B overexpression results in insulin-resistant states; while PTP1B knock-down mice showed enhanced insulin sensitivity in glucose and insulin tolerance, indicating that PTP1B is a major factor in the modulation of insulin sensitivity (Little et al. 2007). In addition, PTP1B is also the key regulator of the leptin signaling pathway. PTP1B-deficient mice also show decreased leptin levels and hypersensitivity to leptin compared with wild-type littermates on low- and high-fat diets (Zabolotny et al. 2002). Despite many drug lead candidates of both natural and synthetic origin, there exist no clinically approved PTP1B inhibitors (Wang et al. 2015). Therefore, immense efforts are being made to identify novel and natural PTP1B inhibitors.
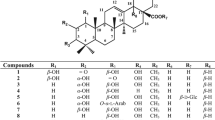
Morus alba (Moraceae), a perennial herb, is the dominant species among the genus Morus. All parts of M. alba (white mulberry) have long been used in traditional Chinese medicine, particularly its leaves, fruits, branches, and root bark, are all recorded in the Chinese Pharmacopoeia (He et al. 2018). In most European countries, ethnobotanical usage of mulberry fruits includes the preparation of juices, wine, jams, marmalades, ice-creams, kinds of vinegar, and cosmetic products (Natić et al. 2015). Traditionally, the leaves of M. alba have been used as herbal tea with important bioactivities such as antioxidant, antidepressant, hypoglycemic, and hepatoprotective effects (Choi et al. 2013; Sánchez-Salcedo et al. 2017). Pharmacological properties of the root bark extract of M. alba include anti-Alzheimer, anti-asthmatic, antibacterial, anti-cancer, anti-depressant, anti-inflammatory, anti-obesity, antioxidant, anti-tyrosinase, cardio-protective, hypoglycaemic, myocardial protective, neuroprotective, and pancreatic lipase inhibitory activities (Yang et al. 2014; Ha et al. 2016, 2018; Kuk et al. 2017). 2-Arylbenzofurans are especially known as the principal constituent of Morus plants and usually comprised of some substituents such as prenyl, geranyl, and farnesyl groups (Fig. 1) (Ha et al. 2016). Among them, farnesylated 2-arylbenzofurans are rarer than prenylated or geranylated 2-arylbenzofurans. The diverse 2-arylbenzofurans result from different positioning of the substituents or cyclization. Previous reports indicate that 2-arylbenzofurans from Morus species have a wide range of biological activities, such as antioxidant, anti-inflammatory, cytotoxicity, and antifungal activities (Yang et al. 2014). Recently, arylbenzofurans isolated from the root bark of M. alba was found to exhibit anti-Alzheimer’s disease and anti-glycation potential (Seong et al. 2018; Paudel et al. 2019). In the previous study, we have reported the isolation of eleven new compounds from M. alba root bark, including seven 2-arylbenzofuran derivatives (morusalfurans A-G), three flavonoids (morusalnols A-C), and one geranylated stilbene (morusibene A), as well as their potential pancreatic lipase (PL) inhibitory activity (Ha et al. 2016). All the farnesylated 2-arylbenzofurans (morusalfurans B and C) showed strong PL inhibitory activity, suggesting the important role of the farnesyl group in the PL inhibitory activity. In the present study, the structure-activity relationship of the above eleven compounds, especially two farnesylated 2-arylbenzofurans in terms of PTP1B inhibitory activity was also investigated. Also, kinetic and molecular docking studies were performed to understand the inhibition type, interaction, and binding mode of the active compounds with the PTP1B enzyme.

Chemical structures of compounds 1–11
Materials and methods
Chemicals and reagents
Ethylenediamine tetraacetic acid (EDTA), p-nitrophenyl phosphate (p-NPP), and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Protein tyrosine phosphatase 1B (PTP1B, human recombinant) and dithiothreitol (DTT) were purchased from Biomol® International LP (Plymouth Meeting, PA, USA) and Bio-Rad Laboratories (Hercules, CA, USA), respectively. All other chemicals and solvents used were of reagent grade and acquired from commercial sources.
Isolation of compounds 1–11 from M. alba root bark
Seven 2-arylbenzofuran derivatives (morusalfurans A–G, 1–7), three flavonoids (morusalnols A–C, 8–10), and one geranylated stilbene (morusibene A, 11) were isolated from the root bark of M. alba in our laboratory as previously described (Ha et al. 2016). The chemical structures of isolated compounds (1–11) are shown in Fig. 1.
PTP1B inhibitory assay
Protein tyrosine phosphatase 1B inhibitory activity was evaluated using p-nitrophenyl phosphate (p-NPP) as the substrate, according to our previously reported procedure with slight modifications (Jung et al. 2013). Sodium orthovanadate and ursolic acid were used as positive controls. In each well of a 96-well plate (final volume 100 µL), 40 µL of PTP1B enzyme [0.5 unit diluted using PTP1B reaction buffer containing 50 mM citrate (pH 6.0), 1 mM ethylenediamine tetraacetic acid (EDTA), 0.1 M NaCl, and 1 mM dithiothreitol (DTT)] was added with or without a test sample. The plate was preincubated at 37 °C for 10 min. After that, 50 µL of 2 mM p-NPP dissolved in PTP1B reaction buffer was added. Following incubation at 37 °C for 15 min in the dark, the reaction was terminated by the addition of 10 µL of 10 M NaOH. The amount of p-nitrophenolate produced by the enzymatic dephosphorylation of p-NPP was estimated by measuring the absorbance at 405 nm using a microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). The non-enzymatic hydrolysis of 2 mM p-NPP was corrected by measuring the increase in absorbance at 405 nm obtained in the absence of the PTP1B enzyme. The percent inhibition (%) was obtained by the following equation:
where Ac is the absorbance of the control, and As is the absorbance of the sample.
Enzyme kinetic analysis with PTP1B
To determine the modes of enzyme inhibition of the active compounds (1−3, 6, 9, and 11), two complementary kinetic methods including Lineweaver-Burk and Dixon plots (Lineweaver and Burk 1934; Dixon 1953) were employed, and Sigma Plot 12.0 was used to analyze and visualize the experimental results (Ha et al. 2018). The enzymatic inhibition of the tested compounds was evaluated by monitoring the effects of different concentrations of the substrates (0.25, 0.50, and 0.75 mM p-NPP for 1 and 3; 0.50, 0.75, and 1.00 mM p-NPP for 2; and 0.37, 0.74, and 1.48 mM p-NPP for 6, 9, and 11) in the Dixon plots (single reciprocal plots). Lineweaver–Burk plots for inhibition of PTP1B were obtained in the presence of various concentrations of the test compounds (0, 5, 10, and 20 µM for 1; 0, 6, 7, and 8 µM for 2; 0, 2, 5, and 7 µM for 3; 0, 10, 15, and 20 µM for 6 and 11; and 0, 20, 25, and 30 µM for 9). The inhibition constants (Ki) were determined by interpretation of Dixon plots, where the value of the x-axis was taken as -Ki.
Molecular docking simulation in PTP1B inhibition
To understand the interaction and binding of the active compounds (1−3, 6, 9, and 11) with PTP1B, molecular docking simulations were performed with AutoDock 4.2 software (Nguyen et al. 2020). The X-ray crystallographic structure of PTP1B (1T49) was obtained from the RCSB Protein Data Bank website (Bernstein et al. 1977; Berman et al. 2002; Wiesmann et al. 2004). The selective allosteric inhibitor A: 3-(3,5-dibromo-4-hydroxy-benzoyl)-2-ethyl-benzofuran-6-sulfonic acid (4-sulfamoyl-phenyl)-amide and selective catalytic inhibitor C: 3-({5-[(N-acetyl-3-{4-[(carboxycarbonyl)(2-carboxyphenyl)amino]-1-naphthyl}-L-alanyl)amino]pentyl}oxy)-2-naphthoic acid were used to detect the respective binding site of the protein. The binding areas of compound C and compound A of the protein were considered to be the most convenient regions for ligand binding in the docking simulation. Protein preparation was conducted using Discovery Studio 2017 R2 software (Accelrys, Inc., San Diego, CA, USA). The 3D structures of compounds 1−3, 6, 9, and 11 were constructed and minimized using Chemdraw and Accelrys Discovery Studio 2017 R2 software. Automated docking simulation was performed using AutoDock Tools (ADT) to assess the appropriate binding orientations and conformations of the ligand molecules with different protein inhibitors. A Lamarckian genetic algorithm method implemented in AutoDock 4.2 was employed. For docking calculations, Gasteiger charges were added by default, the rotatable bonds were set by the AutoDock tools, and all torsions were allowed to rotate. The grid maps were generated by the Autogrid program where the grid box size of 126 × 126 × 126 with a default spacing of 0.375 Å was used. The docking protocol for rigid and flexible ligand docking consisted of 10 independent Genetic Algorithms, while other parameters were used as defaults of the ADT. The binding aspect of PTP1B residues and their corresponding binding affinity score were regarded as the best molecular interaction. The results were analyzed using PyMOL, while the hydrogen bond, van der Waals, and hydrophobic interacting residues were visualized using Discovery studio 2017 R2 software.
Statistics
Statistical significance was analyzed by one-way ANOVA and Duncan’s test (Systat Inc., Evanston, IL, USA), and was noted at p < 0.05. All results are presented as mean ± SEM of at least three independent experiments.
Results
Inhibitory activity against PTP1B
The inhibitory potentials of 1–11 against PTP1B were evaluated using p-nitrophenyl phosphate (p-NPP) as a substrate, and the result was expressed as IC50 values, which were determined by linear regression analysis (Table 1). Specifically, three 2-arylbenzofurans [morusalfurans A–C; (1–3)] showed strong inhibitory effects on PTP1B with IC50 values of 11.02, 8.92, and 7.26 µM, respectively; which are higher compared to the positive controls [sodium orthovanadate (IC50 = 15.10 µM) and ursolic acid (IC50 = 11.34 µM)]. Besides, other 2-arylbenzofuran [morusalfuran F; (6)], one flavonoid [morusalnol B (9)], and one geranylated stilbene [morusibene A (11)] exhibited moderate PTP1B inhibition with IC50 values ranging from 17.64 to 26.56 µM.
The structure-activity relationship (SAR) was observed in the PTP1B inhibitory assay for 2-arylbenzofuran derivatives (1−7). Among them, two farnesylated 2-arylbenzofurans [morusalfurans B (2) and C (3)] showed the strongest inhibitory effect on PTP1B, suggesting the presence of farnesyl group at position 2ʹ positively influenced the PTP1B inhibitory activity of 2-arylbenzofuran derivatives. Interestingly, this suggestion is completely consistent with our previous study, in which the farnesyl group also plays an important role in exerting pancreatic lipase inhibitory activity (Ha et al. 2016).
Enzyme kinetics of PTP1B inhibition
To explain the mode of enzymatic inhibition, the kinetic analysis was performed at different concentrations of the p-NPP substrate and inhibitors (1−3, 6, 9, 11) according to the Lineweaver-Burk plot and Dixon plot methods. The Dixon plot is a graphical method [plot of 1/enzyme velocity (1/V) against inhibitor concentration (I)] employed for the determination of the type of enzyme inhibition and to determine the dissociation or inhibition constant (Ki) for the enzyme-inhibitor complex (Dixon 1953; Cornish-Bowden 1974). In the Lineweaver-Burk plot method, the lines of the inhibitors that intersected at the xy region indicated a mixed inhibition, while the lines which crossed the same point on the x-intercept, represented noncompetitive inhibition (Lineweaver and Burk 1934). As shown in Table 1; Figs. 2 and 3, compounds 1 and 9 showed noncompetitive type inhibition (Ki values; 10.90 and 6.38 µM, respectively), while compounds 2, 3, 6, and 11 showed a mixed type of inhibition (Ki values; 7.09, 6.30, 6.22, and 5.37 µM, respectively) against PTP1B. As the Ki value represents the concentration needed to combine the inhibitor with an enzyme, compounds with a lower Ki value were generally more effective inhibitors against PTP1B, which is an important requisite for the development of preventive and therapeutic agents.

Lineweaver-Burk plots for PTP1B inhibition of 1 (a), 2 (b), and 3 (c). Dixon plots for PTP1B inhibition of 1 (d), 2 (e), and 3 (f)

Lineweaver-Burk plots for PTP1B inhibition of 6 (a), 9 (b), and 11 (c). Dixon plots for PTP1B inhibition of 6 (d), 9 (e), and 11 (f)
Molecular docking simulation of PTP1B inhibition
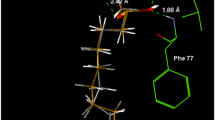
To understand the molecular mechanism and the binding orientation, we further subjected the tested compounds to molecular docking studies using standard software - Autodock 4.2. Our docking simulations were found to be concurrent with the kinetic studies. To validate and optimize the docking procedure, we re-docked a native co-ligand, compound A [3-(3,5-dibromo-4-hydroxy-benzoyl)-2-ethyl-benzofuran-6-sulfonic acid (4-sulfamoyl-phenyl)-amide] into the allosteric site of the PTP1B (PDB ID: 1T49). The root means square deviation (RMSD) value was obtained by comparing the best pose generated in Autodock 4.2 with our docking protocol and the co-ligand (0.56 Å). This validated method was used to investigate the binding pose of the selected compounds. The predicted binding mode of compounds is depicted in Figs. 4, 5 and 6; Table 2. Compounds 2, 3, 6, and 11 demonstrated a mixed type of inhibition (bind to catalytic as well to allosteric site); whereas 1 and 9 showed a non-competitive type of inhibition (allosteric site only). As shown in Fig. 5a and d and Table 2, the oxygen atom at the furan ring and the hydroxyl group at position 3′ of 1 demonstrated interactions with the Asn193 and Lys197 residues via H-bond interaction with a binding energy of – 9.55 kcal/mol. Furthermore, the Gly277, Ser151, Ser190, and Glu200 residues were noted to be involved in the Van der Waals interactions. Also, the benzofuran ring showed π-π stacked hydrophobic interaction with the Phe280 and Leu192 residues while π–σ interaction was observed between CH3-10′′ and Phe196 residue, thus exhibiting similarity with the positive ligand (compound A). In addition, the Ile281, Ala189, Tyr153, and Tyr152 residues were involved in the π-alkyl interactions with 1. In the case of another non-competitive inhibitor, compound 9, no hydrogen bond and π–σ interactions were observed, which explains its slightly less potency than the rest of the active compounds. The Asn193, Lys197, Gly277, Glu276, Pro188, Ser190, and Tyr152 residues were involved in the Van der Waals interactions with 9 while other interactions were similar to 1. As shown in Figs. 4 and 5, compounds 2 and 3 demonstrated binding modes at catalytic as well as at allosteric sites. Both 2 and 3 exhibited binding with the important catalytic residues (Cys215, Tyr46, and Gln262) but with a different binding orientation at the catalytic site with the binding energies of – 6.44 and – 7.14 kcal/mol, respectively (Fig. 4). The OH- and OCH3- group present in the core structure were found to be responsible for the hydrogen bond interactions (Gly220, Gln262, and Gly183 residues with 2; Cys215, Ile219, Thr263 and Gly183 residues with 3) (Fig. 4). However, in the case of 3, additional π–π stacked interaction between Trp179 and ring B was observed. Similarly, the case of the allosteric site also, both 2 and 3 showed interactions with the important allosteric residues including Phe196, Phe280, Leu192, and Asn193 via Van der Waals and hydrophobic interactions, and as expected the orientation was opposite similar as the catalytic pocket (Fig. 5). Only one H-bond interaction was observed between the hydroxyl group at position 6 of 2 and the Lys197 with a binding energy of – 7.13 kcal/mol whereas the most active compound 3, demonstrated two H-bond interactions with Ser187 (OH-6) and Gly277 (OH-5′) with a lowest binding energy of – 9.17 kcal/mol (Fig. 5).

Inhibition mode of 2 (a), 3 (b), 6 (c), and 11 (d) for PTP1B catalytic site with known catalytic inhibitor, compound C (black line). 2D ligand interaction diagram of PTP1B catalytic inhibition by 2 (e), 3 (f), 6 (g), and 11 (h). Dashed lines indicate H-bonds. The figure was generated using PyMOL and Discovery Studio

Inhibition mode of 1 (a), 2 (b), and 3 (c) for PTP1B allosteric site with known allosteric inhibitor, compound A (red line). 2D ligand interaction diagram of PTP1B allosteric inhibition by 1 (d), 2 (e), and 3 (f). Dashed lines indicate H-bonds. The figure was generated using PyMOL and Discovery Studio

Inhibition mode of 6 (a), 9 (b), and 11 (c) for allosteric site with known allosteric inhibitor, compound A (red line). 2D ligand interaction diagram of PTP1B allosteric inhibition by 6 (d), 9 (e), and 11 (f). Dashed lines indicate H-bonds. The figure was generated using PyMOL and Discovery Studio
Furthermore, 6 interacted with Asp48 residue (via H-bond interaction), Val49, Gln262, Ile219, Thr263, Ser216, Gly220, Arg221, Gln266, Gly183, and Ser222 residues (via Van der Waals interactions), and important residues Cys215, Val184, and Trp179 (via π-alkyl interactions) with a binding energy of – 7.26 kcal/mol at the catalytic site (Fig. 4); whereas at the allosteric site, the interactions with Glu276 (via H-bond), Asn193, Ser190, Phe196, Ile281, Gly277, and Ser187 residues (via Van der Waals), Phe280 (via π–π stacked), Leu192, and Ala189 (via π–σ), and Pro188, Tyr152 (via π-Alkyl) were observed (Fig. 6). Interestingly, at both the sites, the hydroxyl group present at the B-ring (OH-3′) was only involved in the hydrogen bond interaction as seen in Figs. 4g and 6d. In addition, compound 11 interacted with Thr263, Glu115, and Lys116 residues (via H-bond interactions), and Gln266, Trp179, Val184, Asp265, Gln262, Ile219, Gly220, Gly218, Asn111, Gly183, Cys215, Lys120, Ala217, and Tyr46 residues (via hydrophobic interactions) with a binding energy of – 6.77 kcal/mol at the catalytic site; whereas in the case of an allosteric site (Fig. 4), the Gly277, and Ser187 residues (via H-bond interactions) and Asn193, Phe196, Ser190, Ala278, Ile281, Phe280, Leu192, Pro188, Ala189, and Glu276 residues (via hydrophobic interactions) were involved in the binding with a binding energy of – 7.67 kcal/mol (Fig. 6). These interactions include various important catalytic and allosteric residues as mentioned earlier, which are essential for proper conformation and the tighter binding, which ultimately leads to better activity. Surprisingly, despite the difference in the structure of 6 and 11, the binding orientation and conformation at the catalytic site as well as at the allosteric site were almost similar.
Discussion
The prevalence of T2DM is increasingly rapidly in an exponential manner and is believed to be the ninth major cause of death. Particularly, the countries with economic transitions towards growth are affected epidemically (Zheng et al. 2018). The current treatment option includes insulin administration and some oral hypoglycemic agents. Chronic use of insulin injection causes suffering and inconvenience to the patients. PTP1B, a negative regulator of the leptin and insulin signaling pathways has been the subject of interest to various researchers as it satisfies the therapeutic target to develop potent and selective inhibitors (Elchebly et al. 1999; Klaman et al. 2000). PTP1B (50 kDa) is composed of 435 amino acid residues and divided into three regions: N-terminal catalytic region, regulatory region (serve as binding sites for proteins and impart substrate specificity), and C-terminal membrane localization region (responsible for the binding the enzyme to the cytoplasmic face of the endoplasmic reticulum). The activity of the enzyme is regulated by phosphorylation of serine and tyrosine residues of catalytic and transmembrane domains. Usually, a shorter version having 298 amino acid residues is employed for enzyme assays. R loop (Val113–Ser118), lysine loop (Leu119–Cys121), WPD loop (Thr177–Pro185), S loop (Ser201–Gly209), Q loop (Ile261–Gln262), α3 helix (Glu186–Glu200), α6 helix (Ala264–Ile281), and α7 helix (Val287–Ser295) play critical roles in dephosphorylation of phosphotyrosine (Shinde et al. 2018). However, due to the lower cell permeability and bioavailability, most of the PTP1B inhibitors are considered as inefficient drug candidates. Also, due to the high polar and conserved nature of the catalytic site of the PTP1B, finding appropriate PTP1B inhibitors remains a harder and big challenge. The first PTP1B inhibitors that entered the clinical trial were ertiprotafib (Wrobel et al. 1999), trodusquemine (Zasloff et al. 2001), and JTT-551(Fukuda et al. 2010). However, ertiprotafib was unsuccessful in Phase II due to its lower potency and undesirable side effects (Zhang and Lee 2003). Until date, there exist no potential PTP1B inhibitors in the market. Consequently, there subsists a greater need for potent PTP1B inhibitors with safe and lesser side effects for the treatment of T2DM. Also, enzymes are highly responsive to the small molecular weight or drug-like molecules and they represent the attractive targets for the drug development and therapy (Mphahlele et al. 2020). Undoubtedly, naturally occurring compounds satisfy the criteria for a potent PTP1B inhibitor. Accordingly, in search of potent natural PTP1B inhibitors, herein, we present the compounds isolated from M. alba root bark, which holds the efficiency to be employed as potential therapeutic agents for T2DM.
All the tested compounds (except 5, 7, and 10) demonstrated the potent inhibitory activity with the IC50 values ranging from 7.26 to 67.86 µM. Among all the compounds, 2 and 3 showed the most potent inhibitory activity against PTP1B. Examination of SARs among the compounds 1–5 suggests that the presence of a farnesyl group at position 2ʹ is essential for potent PTP1B inhibitory activity. Also, various reports regarding the importance of resorcinol scaffold in the structure that increases the efficacy of the compounds in a positive manner (Zhang et al. 2013; Seong et al. 2018). However, in our study, 3 was comparatively more active than 2 (2 has a resorcinol scaffold), which could be due to the interference of the prenyl group at position 6’. Furthermore, considering other similar structures, it can be suggested that the lipophilic farnesyl group is important for strong PTP1B inhibitory activity. Based on our results, it is hypothesized that an increase in the lipophilicity via the addition of prenyl/geranyl/farnesyl moiety at specific positions and a decrease in the charge of aliphatic side chain might be important criteria for enhancement in the inhibitory activity. In addition, kinetics and molecular docking analysis of the selected compounds were done to understand the binding orientation and reason behind the variance in the activity. Comparison of the docking studies between 2 and 3 revealed that 3 exhibited lower binding energy and interacted with the important catalytic residue (Cys215) via H-bond interaction with the hydroxyl group at position 6, which leads to tighter binding at the active site (Fig. 4). Other interactions include binding with the R loop (Glu115 and Lys116), lysine loop (Lys120), WPD loop (Trp179, Pro180, Asp181, Phe182, Gly183, and Val184), Q loop (Gln262), and α6 helix (Asp265 and Gln266). Apart from this, the binding orientation between 2 and 3 was observed to be relatively opposite, which might be due to the presence of additional prenyl moiety at C-6′ position in 2, thus explaining the lower potency of 2 over 3. Similarly, in the case of the allosteric site, 3 exhibited lower binding energy coupled with a greater number of H-bondings, which led to an increase in the binding affinity with better stability and subsequently better activity. Similar results were obtained by Hoang et al. while comparing with the aryl benzofurans against PTP1B (Hoang et al. 2009): mulberrofuran W has the similar structure to that of 3, except for the methoxy group is replaced by hydroxyl group to form resorcinol moiety in 3, and it was found to be three times (IC50; 2.7 µM) more potent than the 3. Likewise, resorcinol moiety present in 1, demonstrated H-bond interaction with the important residues (Asn193 and Lys 197) along with the hydrophobic interactions, which might be the reason behind its profound activity against PTP1B.
Overall, this study reports the first investigation of the PTP1B inhibitory activity of seven 2-arylbenzofuran derivatives (morusalfurans A–G, 1–7), three flavonoids (morusalnols A–C, 8–10), and one geranylated stilbene (morusibene A, 11) isolated from Morus alba root bark. Our results demonstrated two farnesylated 2-arylbenzofurans [morusalfurans B (2) and C (3)] showed strong inhibitory effects on PTP1B, while other 2-arylbenzofurans [morusalfurans A (1) and F (6)], one flavonoid [morusalnol B (9)], and one geranylated stilbene [morusibene A (11)] exhibited moderate PTP1B inhibition. Besides, this study again confirms the importance of the presence of the farnesyl group in the structure of 2-arylbenzofurans for its biological activity. The kinetic studies indicated that morusalfurans B (2), C (3), and F (6) and morusibene A (11) displayed mixed inhibition, while morusalfuran A (1) and morusalnol B (9) displayed noncompetitive inhibition for the PTP1B enzyme. The molecular docking results were in accordance with the kinetic studies and indicated that these active compounds could bind to the active sites of PTP1B with negative binding energies. Coupled with all the evidence that has been published earlier along with our results, it can be concluded that 2-arylbenzofurans are the promising structures for the evaluation as PTP1B inhibitors and should be investigated promptly to develop potent therapeutic agents for T2DM.
References
Alberti KGMM, Zimmet PF (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabet Med 15:539–553. https://doi.org/10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S
Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S (2002) The protein data bank. Acta Crystallogr D Struct Biol 58:899–907. https://doi.org/10.1107/S0907444902003451
Bernstein FC, Koetzle TF, Williams GJB, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M (1977) The protein data bank: a computer-based archival file for macromolecular structures. J Mol Biol 112:535–542. https://doi.org/10.1111/j.1432-1033.1977.tb11885.x
Choi J, Kang HJ, Kim SZ, Kwon TO, Jeong SI, Jang SI (2013) Antioxidant effect of astragalin isolated from the leaves of Morus alba L. against free radical-induced oxidative hemolysis of human red blood cells. Arch Pharm Res 36:912–917. https://doi.org/10.1007/s12272-013-0090-x
Cornish-Bowden A (1974) A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem J 137:143–144. https://doi.org/10.1042/bj1370143
Dixon M (1953) The determination of enzyme inhibitor constants. Biochem J 55:170. https://doi.org/10.1042/bj0550170
Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC (1999) Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283:1544–1548. https://doi.org/10.1126/science.283.5407.1544
Fukuda S, Ohta T, Sakata S, Morinaga H, Ito M, Nakagawa Y, Tanaka M, Matsushita M (2010) Pharmacological profiles of a novel protein tyrosine phosphatase 1B inhibitor, JTT-551. Diabetes Obes Metab 12:299–306. https://doi.org/10.1111/j.1463-1326.2009.01162.x
Ha MT, Tran MH, Ah KJ, Jo K-J, Kim J, Kim WD, Cheon WJ, Woo MH, Ryu SH, Min BS (2016) Potential pancreatic lipase inhibitory activity of phenolic constituents from the root bark of Morus alba L. . Bioorg Med Chem Lett 26:2788–2794. https://doi.org/10.1016/j.bmcl.2016.04.066
Ha MT, Seong SH, Nguyen TD, Cho W-K, Ah KJ, Ma JY, Woo MH, Choi JS, Min BS (2018) Chalcone derivatives from the root bark of Morus alba L. act as inhibitors of PTP1B and α-glucosidase. Phytochemistry 155:114–125. https://doi.org/10.1016/j.phytochem.2018.08.001
He X, Fang J, Ruan Y, Wang X, Sun Y, Wu N, Zhao Z, Chang Y, Ning N, Guo H (2018) Structures, bioactivities and future prospective of polysaccharides from Morus alba (white mulberry): a review. Food Chem 245:899–910. https://doi.org/10.1016/j.foodchem.2017.11.084
Hoang DM, Ngoc TM, Dat NT, Kim YH, Van Luong H, Ahn JS, Bae K (2009) Protein tyrosine phosphatase 1B inhibitors isolated from Morus bombycis. Bioorg Med Chem Lett 19:6759–6761. https://doi.org/10.1016/j.bmcl.2009.09.102
Jung HA, Cho YS, Oh SH, Lee S, Min BS, Moon KH, Choi JS (2013) Kinetics and molecular docking studies of pimarane-type diterpenes as protein tyrosine phosphatase (PTP1B) inhibitors from Aralia continentalis roots. Arch Pharm Res 36:957–965. https://doi.org/10.1007/s12272-013-0131-5
Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim Y-B, Sharpe AH (2000) Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol 20:5479–5489. https://doi.org/10.1128/MCB.20.15.5479-5489.2000
Kuk EB, Jo AR, Oh SI, Sohn HS, Seong SH, Roy A, Choi JS, Jung HA (2017) Anti-Alzheimer’s disease activity of compounds from the root bark of Morus alba L. Arch Pharm Res 40:338–349. https://doi.org/10.1007/s12272-017-0891-4
Lineweaver H, Burk D (1934) The determination of enzyme dissociation constants. J Am Chem Soc 56:658–666. https://doi.org/10.1021/ja01318a036
Little DP, Moran RC, Brenner ED, Stevenson DW (2007) Nuclear genome size in Selaginella. Genome 50:351–356. https://doi.org/10.1139/G06-138
Moller DE (2001) New drug targets for type 2 diabetes and the metabolic syndrome. Nature 414:821–827. https://doi.org/10.1038/414821a
Mphahlele MJ, Choong YS, Maluleka MM, Gildenhuys S (2020) Synthesis, in vitro evaluation and molecular docking of the 5-acetyl-2-aryl-6-hydroxybenzo[b]furans against multiple targets linked to type 2 diabetes. Biomolecules 10:418. https://doi.org/10.3390/biom10030418
Natić MM, Dabić D, Papetti A, Akšić MMF, Ognjanov V, Ljubojević M, Tešić ŽL (2015) Analysis and characterisation of phytochemicals in mulberry (Morus alba L.) fruits grown in Vojvodina, North Serbia. Food Chem 171:128–136. https://doi.org/10.1016/j.foodchem.2014.08.101
Nguyen TTA, Ha MT, Park SE, Choi JS, Min BS, Kim JA (2020) Stilbenes with potent protein tyrosine phosphatase-1b inhibitory activity from the roots of Polygonum multiflorum. J Nat Prod 83:323–332. https://doi.org/10.1021/acs.jnatprod.9b00777
Paudel P, Seong SH, Zhou Y, Ha MT, Min BS, Jung HA, Choi JS (2019) Arylbenzofurans from the root bark of Morus alba as triple inhibitors of cholinesterase, β-site amyloid precursor protein cleaving enzyme 1, and glycogen synthase kinase-3β: relevance to Alzheimer’s disease. ACS Omega 4:6283–6294. https://doi.org/10.1021/acsomega.9b00198
Sánchez-Salcedo EM, Amorós A, Hernández F, Martínez JJ (2017) Physicochemical properties of white (Morus alba) and black (Morus nigra) mulberry leaves, a new food supplement. J Food Nutr Res 5:253–261. https://doi.org/10.12691/jfnr-5-4-7
Seong SH, Ha MT, Min BS, Jung HA, Choi JS (2018) Moracin derivatives from Morus Radix as dual BACE1 and cholinesterase inhibitors with antioxidant and anti-glycation capacities. Life Sci 210:20–28. https://doi.org/10.1016/j.lfs.2018.08.060
Shinde RN, Kumar GS, Eqbal S, Sobhia ME (2018) Screening and identification of potential PTP1B allosteric inhibitors using in silico and in vitro approaches. PLoS ONE 13:e0199020. https://doi.org/10.1371/journal.pone.0199020
Wang LJ, Jiang B, Wu N, Wang SY, Shi DY (2015) Small molecules as potent protein tyrosine phosphatase 1B (PTP1B) inhibitors documented in patents from 2009 to 2013. Mini Rev Med Chem 15:104–122. https://doi.org/10.2174/1389557515666150203144339
Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, Fahr BJ, Zhong M, Taylor L, Randal M, Mcdowell RS, Hansen SK (2004) Allosteric inhibition of protein tyrosine phosphatase 1B. Nat Struct Mol Biol 11:730–737. https://doi.org/10.1038/nsmb803
World Health Organization (2016) Global report on diabetes. https://apps.who.int/iris/bitstream/handle/10665/204871/9789241565257_eng.pdf. Accessed 7 Apr 2016
Wrobel J, Sredy J, Moxham C, Dietrich A, Li Z, Sawicki DR, Seestaller L, Wu L, Katz A, Sullivan D (1999) PTP1B inhibition and antihyperglycemic activity in the ob/ob mouse model of novel 11-arylbenzo[b]naphtho [2,3-d] furans and 11-arylbenzo[b]naphtho [2,3-d] thiophenes. J Med Chem 42:3199–3202. https://doi.org/10.1021/jm990260v
Yang Y, Tan Y-X, Chen R-Y, Kang J (2014) The latest review on the polyphenols and their bioactivities of Chinese Morus plants. J Asian Nat Prod Res 16:690–702. https://doi.org/10.1080/10286020.2014.923405
Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim Y-B, Elmquist JK, Tartaglia LA, Kahn BB (2002) PTP1B regulates leptin signal transduction in vivo. Dev Cell 2:489–495. https://doi.org/10.1016/S1534-5807(02)00148-X
Zasloff M, Williams J, Chen Q, Anderson M, Maeder T, Holroyd K, Jones S, Kinney W, Cheshire K, Mclane M (2001) A spermine-coupled cholesterol metabolite from the shark with potent appetite suppressant and antidiabetic properties. Int J Obes (Lond) 25:689–697. https://doi.org/10.1038/sj.ijo.0801599
Zhang ZY, Lee SY (2003) PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. Expert Opin Investig Drugs 12:223–233. https://doi.org/10.1517/13543784.12.2.223
Zhang SS, Ma QY, Zou XS, Dai HF, Huang SZ, Luo Y, Yu ZF, Luo HR, Zhao YX (2013) Chemical constituents from the fungus Amauroderma amoiensis and their in vitro acetylcholinesterase inhibitory activities. Planta Med 79:87–91. https://doi.org/10.1055/s-0032-1327951
Zheng Y, Ley SH, Hu FB (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 14:88–98. https://doi.org/10.1038/nrendo.2017.151
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2020R1F1A1072001) and the Ministry of Education (2016R1D1A1B03930246), Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ha, M.T., Shrestha, S., Tran, T.H. et al. Inhibition of PTP1B by farnesylated 2-arylbenzofurans isolated from Morus alba root bark: unraveling the mechanism of inhibition based on in vitro and in silico studies. Arch. Pharm. Res. 43, 961–975 (2020). https://doi.org/10.1007/s12272-020-01269-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-020-01269-4