
Abstract
The somatic mutation of liver kinase B1 (LKB1) has been implicated in various tumors, which is reflected in the survival, proliferation, and metastasis of tumor cells. However, the regulation of LKB1 in lipid metabolism, a process that is involved in tumor progression is not completely clear. We conclude that LKB1 deficiency results in abnormal expression and activation of multiple molecules related to lipid metabolism which locate downstream of AMP-activated protein kinase (AMPK) or salt-induced kinase (SIK). Abnormal lipid metabolism induced by LKB1 deficiency contributes to the proliferation and metastasis of tumor cells through energy regulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liver Kinase B1 (LKB1), also known as the serine-threonine kinase (STK) 11, is a tumor suppressor gene. LKB1 functions as a protein kinase which plays a vital role in maintaining energy homeostasis by phosphorylating and activating AMP-activated protein kinase (AMPK) family members [1]. LKB1 mutation is associated with the occurrence, development, and poor prognosis of several types of cancer, such as lung cancer, melanoma, cervical cancer, and hepatocellular carcinoma (HCC) [2,3,4,5].
Proliferation and metastasis of tumor cells involved in alteration of energy metabolism [6]. Although glucose and glutamine are important sources to maintain energy requirements for tumor cells [7], fatty acid oxidation (FAO) also provides potent metabolism support and possesses higher energy conversion efficiency [8]. In addition, excessive accumulation of lipid, which is stored in the form of lipid droplets, maintains the survival of tumor cells in energy-restricted condition [9]. LKB1 participates in the regulation of lipid synthesis, oxidation, and uptake in two ways. On the one hand, LKB1 activates downstream kinases, such as salt-induced kinase (SIK) and AMPK, which involve the activation and expression of several proteins related to lipid metabolism [10]. For instance, multiple key enzymes and transcription factors locate downstream of LKB1 and participate in de novo FA synthesis, such as sterol regulatory element-binding protein (SREBP), acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), and stearoyl-CoA desaturase-1 (SCD1) [11]. Upregulation of these FA synthesis enzymes was displayed promoting the development of several types of human cancer [12]. On the other hand, LKB1 regulates glucose metabolism via direct or indirect manners, such as the pentose phosphate pathway (PPP), which provides NADPH to lipid synthesis [13, 14]. Therefore, we focused on pathways under the regulation of LKB1 which involve lipid metabolism, and analyzed their role in the proliferation or metastasis of tumor cells. We are certain that the development and application of targeted inhibitors that process function to reverse abnormal lipid metabolism will contribute to improving prognosis in LKB1-deficient tumors.
Lipid metabolism pathways under the regulation of LKB1
LKB1-AMPK pathway
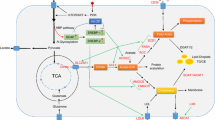
AMPK is an uppermost downstream kinase of LKB1, which functions as a central regulator of energy metabolism [15]. AMPK plays a vital role in energy metabolism and maintaining physiological functions in multiple types of cells and tissues (Fig. 1). For instance, AMPK mediates LKB1 deletion-induced enhancement of lipid-related gene expression in proliferative myoblasts [16]. Activation of AMPK significantly reduces hepatic triglyceride (TG) levels and stimulates the utilization of FA through restoring FAO [17] and inhibiting the de novo synthesis of lipid and cholesterol [18]. Activation of the LKB1-AMPK pathway also facilitates hepatocyte autophagy, which reduces lipotoxicity induced by lipid accumulation by blocking the formation of lipid droplets (LDs) [19]. Moreover, the AMPK-activated insulin-induced gene (Insig) is a protein that is bound with oxysterol in the endoplasmic reticulum (ER), which suppresses FA synthesis through inhibiting SREBP1c cleavage [20]. Liraglutide (LRG) is an agonist of Glucagon-like peptide-1 (GLP-1) receptor, which inhibits lipid synthesis by activating the AMPK-mechanistic target of rapamycin complex 1 (mTORC1)-SREBP1 pathway to ameliorate hepatic lipid accumulation [21].

AMPK regulates lipid metabolism in multiple tissues and cells. Activation of AMPK reduces lipid accumulation in the liver, adipocyte, islet cell, tumor cell, and myoblast via modulating the expression of genes related to lipid metabolism, inhibiting activities of metabolic enzymes involved in lipid biosynthesis, and intensifying FAO and autophagy. AMPK AMP-activated protein kinase; SREBP-1 sterol regulatory element-binding protein 1; ACC acetyl-CoA carboxylase; FASN fatty acid synthase; TG triglyceride; FAO fatty acid oxidation; Insig insulin-induced gene; GLP-1 glucagon-like peptide-1; mTOR mechanistic target of rapamycin; ACLY ATP citrate lyase; CRTC2 cAMP-regulated transcriptional coactivator 2; CREB cAMP response element binding protein
AMPK negatively regulates ACC which mediates adipogenesis by catalyzing the formation of malonyl-CoA [22]. AMPK activation reduces the expression of FASN and ATP-citrate lyase (ACLY) and inhibits the activity of SREBP1, which suppresses tumor development in breast and prostate cancer [23, 24]. Together, AMPK regulates lipid metabolism by inhibiting lipid synthesis and promoting FAO, and AMPK deficiency-induced excessive lipid accumulation provides sufficient energy for the proliferation and metastasis of tumor cells. Notably, AMPK is also regulated by the ratio of AMP/ATP and calcium/calmodulin-dependent protein kinase levels [25, 26] Although LKB1 is responsible for regulating AMPK activation as the main upstream kinase, other signal pathways need to be considered when studying LKB1-AMPK pathway.
LKB1-SIK pathway
SIKs are members of the AMPK superfamily, which functions through controlling phosphorylation and the subcellular localization of class IIa histone deacetylases (IIa HDACs) and cAMP-regulated transcriptional coactivators (CRTCs) [10]. Activation of the LKB1-SIK pathway suppresses tumor development, which had been confirmed in a mouse model of lung adenocarcinoma [27]. There were 38% upregulated genes which can be attributed to SIK1/3 inactivation in all upregulated genes induced by LKB1 deficiency in non-small cell lung cancer (NSCLC) [27]. Besides, LKB1-SIK3-HDAC4 signaling functions to repair the lipid solubility defect induced by LKB1 mutants in drosophila, which is a SIK3-dependent process that isn’t affected by activation of AMPK [28]. Further study suggested that LKB1-SIK3-HDAC4 had been regarded as a potential therapeutic target for acute myeloid leukemia [29].
LKB1-SIK2 regulates the phosphorylation of CRTC2, CRTC3, and HDAC4 in adipocytes [30]. Deletion of CRTC2 reduces the expression of SREBP1c, FASN, and ACC [30]. SIK2 not only determines the properties of white adipose tissue by controlling the activity of cAMP-response element-binding protein (CREB)-CRTC2-dependent transcription [31], but also promotes cholesterol synthesis by upregulating the expression of SREBP2 in ovarian cancer cells [32]. We consider that LKB1-SIK-CRTC/HDAC4 may act as a potential target for the inhibition of tumor growth.
PPP affects lipid synthesis by providing NADPH
PPP produces ribulose-5-phosphate (Ru-5-P) and nicotinamide adenine dinucleotide phosphate (NADPH) [13]. Glucose-6-phosphate dehydrogenase (G6PD) is a rate-limiting enzyme of PPP, which is an important target of LKB1 [14]. Thus, LKB1 deficiency-induced upregulation of lipid synthesis can be attributed to intensifying PPP which provides material and facilitate the expression of genes related to FA synthesis.
PPP flux mediated by LKB1-AMPK-HDAC10-G6DP signaling had been shown to promote the growth of lung cancer cells [14]. However, LKB1-AMPK signaling is activated in breast cancer which reduces the level of G6PD transcription [33]. We speculated that upstream stimulators and tumor backgrounds underlie the different regulatory effects of LKB1 on G6PD. In the presence of glycolysis disorder, the levels of NADPH in LKB1-mutant A549 cells are higher compared to H1334 cells with normal LKB1 [34]. Nonetheless, Ru-5-P level was not changed in A549 cells with LKB1 overexpression or H1299 cell with LKB1 knocked down [14], which provides evidence that LKB1 regulates NADPH level through G6PD but Ru-5-P might be interfered with by other pathways. In addition, activation of the AMPK-SIRT1-p53 pathway increases the phosphorylation of ACC in A549 and NCI-H1299 cells [35], while downregulation of p53 increases G6PD activity in colon cancer [36], which leads us to establish a relationship between LKB1-AMPK signaling and p53-mediated G6PD regulation. Thus, further studies on the connection between LKB1, PPP, and tumor lipid metabolism are needed.
CD36 promotes lipid uptake and storage
CD36 is an important FA transporter located on the surface of various types of cells. LKB1 deficiency results in increased expression of CD36 and decreased expression of ATP binding cassette (ABC) transporter, which leads to dysfunction of cholesterol uptake, de novo synthesis, and outflow [37]. In lapatinib-resistant breast cancer cells, a gene set enrichment analysis (GSEA) and Gene Ontology (GO) enrichment analysis revealed a significant enrichment of lipid-related pathways [38]. However, upregulated of CD36 was principal, and there were no significant differences in FASN and carnitine palmitoyltransferase-1A (CPT1A) levels [38], which provides a hypothesis that CD36 mediated lipid uptake and storage, but not lipid synthesis of FAO, plays an irreplaceable role in metastatic of breast cancer cells. Deletion or inhibition of CD36 limits the uptake of FAs from the tumor microenvironment and reduces adipocyte-mediated invasion and migration of prostate cancer cells [39]. In addition, CD36 is closely related to the survival and metastasis of numerous tumors, such as lung cancer, bladder cancer, breast cancer, and melanoma [40]. Therefore, improvement of disorder lipid metabolism by blocking CD36 is an effective therapeutic strategy for inhibiting tumor development, which deserves to be expected in clinical transformation.
Other downstream lipid regulators of LKB1
Although regulation of lipid metabolism by LKB1 is a complex process (Fig. 2), energy storage is the ultimate purpose which is provided to sustain survival under energy stress conditions. Thus, LKB1 deficiency-induced abnormal lipid metabolism is displayed as enhancement in FA uptake and FA synthesis, and suppression in FAO. The LKB1-mTOR signaling inhibits differentiation of adipocyte and TG accumulation in brown adipose tissue by activating FAO and downregulating peroxisome proliferator-activated receptor γ (PPARγ) [41, 42]. Serine/arginine-rich protein kinase 2 (SRPK2) is required for de novo lipogenesis, which is under the regulation of mTOR [43]. Acetyl-CoA mediates a negative regulatory loop in the LKB1-AMPK-ACC pathway, which is a substrate of FA synthesis but participates in LKB1 acetylation [44]. We summarized that LKB1 mediated regulation of lipid metabolism processes complexity, further experimental investigations need to be conducted in different types of cancers because of the varied genetic background.

Pathway mediated regulation of LKB1 in lipid metabolism. LKB1 inhibits SREBP-induced lipid synthesis and facilitates fatty oxidation through activating AMPK. LKB1 also phosphorylates HDAC and CRTC via activating SIKs, which further inhibit the expression of lipogenic genes. LKB1 reduces lipid uptake by regulating lipid membrane transporters. LKB1 facilitates and suppresses lipid synthesis via upregulating and downregulating PPP in tumor cells with different background genotypes. LKB1 liver kinase B1, AMPK AMP-activated protein kinase, SIK salt-induced kinase, mTOR mechanistic target of rapamycin, ACC acetyl-CoA carboxylase, FAO fatty acid oxidation, SREBP sterol regulatory element-binding protein, FASN fatty acid synthase, SCD1 stearoyl-CoA desaturase 1, ACLY ATP citrate lyase, PPP pentose phosphate pathway, G6PD glucose-6-phosphate dehydrogenase, NADPH nicotinamide adenine dinucleotide phosphate, SRPK2 serine/arginine-rich protein kinase 2, HDAC4 histone deacetylase 4, CRTC2 cAMP-regulated transcriptional coactivator 2, CREB cAMP response element-binding protein, ABC ATP binding cassette, CPT1 carnitine palmitoyltransferase 1
Upstream regulated molecules of the LKB1 pathway
SIRT
Sirtuin 1 (SIRT1) is required for AMPK activity. Bouchardatine is an effective inhibitor of adipogenesis, which reprograms lipid metabolism by activating SIRT1-AMPKα-ACC in rectal cancer [45], further study revealed that LKB1 mediates this signal transduction process [46, 47]. Notably, SIRT1 promotes adipocyte lipolysis in mesenteric adipose tissue but has the opposite inhibitory effect in epididymal adipose tissue [48], which illustrates certain restricted conditions, such as tissue types or genetic background, affect the role of SIRT1 in the regulation of lipid metabolism. Even so, we considered that the targeted SIRT1-LKB1 pathway remains a potential therapeutic strategy for tumors with explicit genetic background.
cAMP/PKA
cAMP-dependent protein kinase A (PKA) functions transcriptional regulation of genes related to adipogenesis. The melanocortin system plays a key role in controlling appetite and adipogenesis [49]. An investigation showed a pathway that α-Melanocyte-stimulating hormone (α-MSH)-cAMP-PKA-extracellular signal-regulated kinase (ERK) -LKB1-AMPK in hypothalamus cells, which provides a therapeutic target for lipid metabolic diseases [50]. In addition, cAMP also activates the LKB1-AMPK-SREBP pathway, which suggested that increased cAMP regulates lipid metabolism by reducing SREBPs expression [51].
PKA-LKB1 signaling pathway inhibits 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), ACC, and SREBP-1 through activating AMPK in insulin-resistant HepG2 cells [52]. Although LKB1 has been considered a tumor suppressor traditionally, it functions as the facilitation of tumorigenesis in thyroid cancer as the downstream mediator of PKA [53]. On the one hand, PKA opposes the negative regulation of lipolysis through phosphorylating AMPKα1 at ser173 which inhibits the activation of AMPK mediated by LKB1. On the other hand, activation of PKA directly stimulates adipocyte lipolysis, which triggers a negative feedback mechanism of AMPK. The appropriate utilization of the negative feedback loop balance between PKA and AMPK contributes to the precise regulation of lipolysis response [54]. The complex regulatory mechanism between PKA, LKB1, and AMPK provides support for maintaining metabolic balance, and may also promote tumor development, which needs to further study.
Hormones and inflammation
Hormones and inflammation are important factors that affect the development of cancers. Given that aromatase mediates hormone synthesis and several inflammatory mediators are classed as lipid, abnormal lipid metabolism also participates in the development of tumors through regulating hormones and inflammation. Prostaglandin E-2 (PGE2), an inflammatory mediator, inhibits the activity of the LKB1-AMPK pathway and regulates aromatase in breast adipose stromal cells [55]. In addition, it has been indicated that leptin induces aromatase expression by inhibiting the LKB1-AMPK pathway and subsequently nuclear translocation of CRTC2, while adiponectin has the opposite effect to leptin [56]. Vitamin D has been found to inhibit aromatase expression and local estrogen synthesis in tumor cells and adipose tissues through increasing LKB1 activity in response to the adverse effect of obesity that contributes to breast cancer growth [57]. In conclusion, the targeted LKB1 pathway may provide effective intervention for the high-risk obese people who need to prevent the occurrence and development of breast cancer more than others.
LKB1 deficiency-induced abnormal metabolism facilitates tumor progress
ACC-driven lipogenesis supports cell survival and metastasis of tumors
ACC is a major downstream lipid regulator of LKB1-AMPK signaling. ACC accelerates FAO in two CPT1-dependent manners. On the one hand, phosphorylation of ACC inactivates the ability to mediate malonyl-coA production, which promotes FAO with increased CPT1A content [58]. On the other hand, the formation of a complex between ACCα and CPT1A attenuates in the case of glucose deficiency, which boosts FAO by facilitating the transport of FA [59]. Upregulation of ACCα and p-ACC are predictors of recurrence and poor survival of tumors [59, 60]. Thus, we considered that ACC has the potential to act as a detection index, a tumor prognostic marker, or a therapeutic target.
SREBP1 maintains lipid synthesis and tumor cell viability
SREBP locates downstream of LKB1, and AMPK or SIK mediates the regulation of LKB1 for SREBPs. Activation of SREBPs regulates the expression of genes related to the key enzymes in cholesterol synthesis and FA synthesis [12]. Multiple types of tumors display activation of the mTORC1-SREBP pathway, which facilitates lipid synthesis [61]. In addition, SREBP1 is essential for the viability of cancer cells under hypoxic conditions [62]. SREBP1 is associated with the degree of malignancy and prognosis of tumors, which has been identified in metastatic melanoma [63]. Inhibition of SREBP1 increases the sensitivity of tumor therapy [64, 65], which inspires us to further develop selective SREBP1 inhibitors to suppress the development of tumors.
SCD promotes tumor growth and proliferation
SCD supports lipogenesis and desaturation of tumor cells under the reduced supply of exogenous lipids, which is important for cell viability and proliferation of tumors [66]. In addition, SCD expression is related to the survival time of patients, which can be used as a marker of tumor prognosis [67, 68]. The expression level of SCD1 in lung adenocarcinoma was higher than in adjacent normal tissues [67]. High expression of SCD usually indicates the presence of advanced disease in breast cancer and prostate cancer [66]. SCD1 also promotes the migration and invasion of tumor cells [67]. Thus, targeted inhibition of SCD has the potential to improve prognosis in tumors with high SCD expression.
Promoting effect of CPT1A on cancer
CPT1 is a rate-limiting enzyme of FAO, whose isozyme CPT1A deficiency inhibits the invasion and growth of radiotherapy-resistant breast cancer by reducing FAO [69]. A gene transcriptional splicing variant CPT1Av2 expressed in breast cancer cells interacts with HDAC1 molecules, which regulates the epigenetic inheritance of genes involved in tumor-associated cell death and invasion pathways [70]. In addition, the anti-tumor mechanism of mTORC involves the reduction of CPT1A expression and lipid catabolism [71]. Down-regulated CPT1A expression reduces lipid utilization by attenuating lipid catabolism, which contributes to the downregulation of cancer-related gene expression and apoptosis of the tumor cells [69,70,71]. These results revealed that targeting FAO contributes to the regulation of tumor energy metabolism, and CPT1A is a potential metabolic target in cancer therapy.
ATGL affects tumor energy storage
Adipose triglyceride lipase (ATGL) is a rate-limiting enzyme in the triglyceride hydrolysis cascade, whose expression is reduced in malignant tumors [72]. The absence of ATGL induces the development of lung tumors in animal models and low levels of ATGL are associated with poor survival in patients with ovarian cancer, NSCLC, breast cancer, and stomach cancer [72]. In addition, the lipolysis activity of ATGL is crucial for the distant metastasis of tumor cells, which depends on the gradual release of stored free FA by LDs [73]. ATGL is required for the adipocyte-mediated proliferation of breast cancer cells, which uptake energy from adipocytes through the transfer of fatty acids [74]. Taken together, ATGL is considered to exert a tumor-inhibiting effect due to its deficiency in malignant tumors. However, as mechanisms of lipid metastasis provide great energy support for tumor cells, ATGL involves in tumor aggressiveness, which demonstrates further studies are required to confirm the effects of ATGL on tumor growth.
Application: targeted inhibitors of LKB1 pathways on liver diseases and tumors
Inhibitors activate the LKB1 lipid regulatory pathways
The tankyrase antagonist G007-LK was shown to play a dual role in lowering blood glucose and inhibiting the development of lung cancer by activating the LKB1-AMPK pathway [75]. A pan-Pim kinase antagonist AZD1208 with anti-cancer activity enhances LKB1 phosphorylation and reduces p-ACC, which resists adipogenesis [76]. ONC201 is an antagonist of dopamine receptor D2, which exerts anti-tumor effects in both obese and non-obese LKB1fl/fl/p53fl/fl mice with endometrial tumors. Mechanically, results from metabonomic analyses showed that ONC201 upregulated lipid synthesis [77]. Honokiol is a potential leptin antagonist, which induces SIRT1/3-mediated activation of LKB1-miR-34a signaling to antagonize tumorigenesis induced by leptin in breast cancer [78].
Activators target LKB1 downstream to release liver diseases
Several types of liver diseases relate to abnormal lipid metabolism [79]. We summarized the active factors of LKB1 and possible therapeutic effects in lipid metabolism disorders in liver diseases (Table 1). Notably, natural plant extracts and their derived compounds are beneficial to correct metabolic disorders, which applicated may exert energy stress on tumor cells to achieve suppressed effect.
Inhibitors target LKB1 downstream to treat tumors
Several targeted inhibitors of the LKB1 pathway which relate to lipid regulation have been conducted in extensive preclinical trials in tumors. ND646, an ACC allosteric inhibitor, exerted a strong inhibitory effect on NSCLC [86]. Selective SREBP1 inhibitors reduce the expression of FASN, HMGCR, and SCD1, which decrease lipid synthesis in pancreatic cancer cells [87]. Sorafenib targets SCD1 through the ATP-AMPK-mTOR-SREBP1 signaling pathway and induces the death of HCC cells [88]. CPT1 inhibitor ST1326 counteracts the proliferation of chronic lymphocytic leukemia cells by blocking FAO and depleting acetyl-CoA in the cytoplasm [89].
Drug resistance is a practical problem in tumor treatment. ACC mutants with AMPK phosphorylation site deficiency were reported to protect head and neck squamous cell carcinoma cells to escape cetuximab-induced growth inhibition [90]. The incidence of BRAF gene mutations in melanoma is as high as 40–50%, but the therapeutic effect is not optimal when targeted BRAF. Owing to SREBP1-dependent continuous lipogenesis being a key mechanism of drug resistance induced by the BRAF mutant, simultaneous inhibition of SREBP1 enhances the sensitivity of melanoma [91], which suggests that identifying the mechanism of resistance and then synergistic use of agents will better sensitize and combat tumors.
Conclusion
LKB1 demonstrates a satisfactory ability to inhibit tumor progression, whose mutation is a mark of poor prognosis of tumor. Different cell sources and genetic backgrounds affect the expression and function of LKB1. However, LKB1 deficiency-induced abnormal lipid metabolism which is mediated by the disordering of transcriptional regulatory factors was proved to be detrimental. This review focuses on the role of LKB1-mediated pathways, which affect tumor progression through disturbing lipid metabolism. Because of the drug resistance of existing inhibitors, bimolecular targeted inhibitors and chemotherapy or radiotherapy combined with targeted inhibitors improve the therapeutic effect of tumors. In-depth studies of LKB1 are contributed to combining multiple targets for detection, which improves the accuracy of tumor hierarchical classification and stratification based on LKB1 expression. We considered that the selection of appropriate target inhibitors for sensitive tumor types may improve malignant lipid metabolism, which has the potential to inhibit tumor proliferation and metastasis.
Abbreviations
- LKB1:
-
Liver kinase B1
- STK:
-
Serine-threonine kinase
- HCC:
-
Hepatocellular carcinoma
- FA:
-
Fatty acid
- AMPK:
-
AMP-activated protein kinase
- SIK:
-
Salt-induced kinase
- PPP:
-
Pentose phosphate pathway
- SREBP:
-
Sterol regulatory element-binding protein
- ACC:
-
Acetyl-CoA carboxylase
- FASN:
-
Fatty acid synthase
- SCD1:
-
Stearoyl-CoA desaturase 1
- TG:
-
Triglyceride
- FAO:
-
Fatty acid oxidation
- LDs:
-
Lipid droplets
- Insig:
-
Insulin-induced gene
- ER:
-
Endoplasmic reticulum
- GLP-1:
-
Glucagon-like peptide-1
- LRG:
-
Liraglutide
- mTORC1:
-
Mechanistic target of rapamycin complex 1
- KO:
-
Knockout
- ACLY:
-
ATP citrate lyase
- HDACs:
-
Histone deacetylases
- CRTC:
-
CAMP-regulated transcriptional coactivator
- CREB:
-
CAMP response element binding protein
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- Ru-5-P:
-
Ribulose-5-phosphate
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- ABC:
-
ATP binding cassette
- GSEA:
-
Gene set enrichment analysis
- GO:
-
Gene Ontology
- CPT1A:
-
Carnitine palmitoyltransferase 1A
- PPAR:
-
Peroxisome proliferator-activated receptor
- SRPK2:
-
Serine/arginine-rich protein kinase 2
- ATGL:
-
Adipose triglyceride lipase
- NSCLC:
-
Non-small cell lung cancer
- SIRT1:
-
Sirtuin 1
- NAFLD:
-
Non-alcoholic fatty liver disease
- PKA:
-
CAMP-dependent protein kinase A
- ERK:
-
Extracellular signal-regulated kinase
- HMGCR:
-
3-Hydroxy-3-methylglutaryl-CoA reductase
- HSF1:
-
Heat shock factor 1
- PGE2:
-
Prostaglandin E2
References
Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18(1):38–43.
Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–10.
Zheng B, Jeong JH, Asara JM, et al. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009;33(2):237–47.
Zeng Q, Chen J, Li Y, et al. LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene. 2017;36(9):1245–55.
Wu CC, Wu DW, Lin YY, Lin PL, Lee H. Hepatitis B virus X protein represses LKB1 expression to promote tumor progression and poor postoperative outcome in hepatocellular carcinoma. Surgery. 2018;163(5):1040–6.
Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s achilles’ heel. Cancer Cell. 2008;13(6):472–82.
Zhu L, Zhu X, Wu Y. Effects of glucose metabolism, lipid metabolism, and glutamine metabolism on tumor microenvironment and clinical implications. Biomolecules. 2022;12(4):580.
Ma Y, Temkin SM, Hawkridge AM, et al. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. 2018;435:92–100.
Koizume S, Miyagi Y. Lipid droplets: a key cellular organelle associated with cancer cell survival under normoxia and hypoxia. Int J Mol Sci. 2016;17(9):1430.
Gormand A, Berggreen C, Amar L, et al. LKB1 signalling attenuates early events of adipogenesis and responds to adipogenic cues. J Mol Endocrinol. 2014;53(1):117–30.
Seo MS, Kim JH, Kim HJ, Chang KC, Park SW. Honokiol activates the LKB1-AMPK signaling pathway and attenuates the lipid accumulation in hepatocytes. Toxicol Appl Pharmacol. 2015;284(2):113–24.
Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38(1):27.
Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39(8):347–54.
Shan C, Lu Z, Li Z, et al. 4-hydroxyphenylpyruvate dioxygenase promotes lung cancer growth via pentose phosphate pathway (PPP) flux mediated by LKB1-AMPK/HDAC10/G6PD axis. Cell Death Dis. 2019;10(7):525.
Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31–7.
Shan T, Zhang P, Bi P, Kuang S. Lkb1 deletion promotes ectopic lipid accumulation in muscle progenitor cells and mature muscles. J Cell Physiol. 2015;230(5):1033–41.
Boudaba N, Marion A, Huet C, Pierre R, Viollet B, Foretz M. AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine. 2018;28:194–209.
Esquejo RM, Salatto CT, Delmore J, et al. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine. 2018;31:122–32.
Huang Y, Liu F, Zhang F, Liu P, Xu T, Ding W. Vanadium(IV)-chlorodipicolinate alleviates hepatic lipid accumulation by inducing autophagy via the LKB1/AMPK signaling pathway in vitro and in vivo. J Inorg Biochem. 2018;183:66–76.
Han Y, Hu Z, Cui A, et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat Commun. 2019;10(1):623.
Hao T, Chen H, Wu S, Tian H. LRG ameliorates steatohepatitis by activating the AMPK/mTOR/SREBP1 signaling pathway in C57BL/6J mice fed a high-fat diet. Mol Med Rep. 2019;20(1):701–8.
Thomson DM, Brown JD, Fillmore N, et al. LKB1 and the regulation of malonyl-CoA and fatty acid oxidation in muscle. Am J Physiol Endocrinol Metab. 2007;293(6):E1572–9.
Zhang ZG, Zhang HS, Sun HL, Liu HY, Liu MY, Zhou Z. KDM5B promotes breast cancer cell proliferation and migration via AMPK-mediated lipid metabolism reprogramming. Exp Cell Res. 2019;379(2):182–90.
Zheng Y, ** J, Gao Y, Luo C, Wu X, Liu J. Phospholipase Cε regulates prostate cancer lipid metabolism and proliferation by targeting AMP-activated protein kinase (AMPK)/sterol regulatory element-binding protein 1 (SREBP-1) signaling pathway. Med Sci Monit. 2020;26: e924328.
Stenesen D, Suh JM, Seo J, et al. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 2013;17(1):101–12.
Wang MD, Wu H, Huang S, et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget. 2016;7(6):6711–26.
Hollstein PE, Eichner LJ, Brun SN, et al. The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer Discov. 2019;9(11):1606–27.
Choi S, Lim DS, Chung J. Feeding and fasting signals converge on the LKB1-SIK3 pathway to regulate lipid metabolism in drosophila. PLoS Genet. 2015;11(5): e1005263.
Tarumoto Y, Lu B, Somerville TDD, et al. LKB1, salt-inducible kinases, and MEF2C are linked dependencies in acute myeloid leukemia. Mol Cell. 2018;69(6):1017–27.
Henriksson E, Säll J, Gormand A, et al. SIK2 regulates CRTCs, HDAC4 and glucose uptake in adipocytes. J Cell Sci. 2015;128(3):472–86.
Park J, Yoon YS, Han HS, et al. SIK2 is critical in the regulation of lipid homeostasis and adipogenesis in vivo. Diabetes. 2014;63(11):3659–73.
Zhao J, Zhang X, Gao T, et al. SIK2 enhances synthesis of fatty acid and cholesterol in ovarian cancer cells and tumor growth through PI3K/Akt signaling pathway. Cell Death Dis. 2020;11(1):25.
Yang L, He Z, Yao J, et al. Regulation of AMPK-related glycolipid metabolism imbalances redox homeostasis and inhibits anchorage independent growth in human breast cancer cells [published correction appears in Redox Biol. 2020 Jan; 28:101382]. Redox Biol. 2018;17:180–91.
**ao Y, Kwong M, Daemen A, et al. Metabolic response to NAD depletion across cell lines is highly variable. PLoS ONE. 2016;11(10): e0164166.
Zhang Y, Sun C, **ao G, et al. S-nitrosylation of the peroxiredoxin-2 promotes S-nitrosoglutathione-mediated lung cancer cells apoptosis via AMPK-SIRT1 pathway. Cell Death Dis. 2019;10(5):329.
Zhang X, Zhang X, Li Y, et al. PAK4 regulates G6PD activity by p53 degradation involving colon cancer cell growth. Cell Death Dis. 2017;8(5): e2820.
Timilshina M, You Z, Lacher SM, et al. Activation of mevalonate pathway via LKB1 is essential for stability of treg cells. Cell Rep. 2019;27(10):2948-2961.e7.
Feng WW, Wilkins O, Bang S, et al. CD36-mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies. Cell Rep. 2019;29(11):3405–20.
Watt MJ, Clark AK, Selth LA, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med. 2019;11(478):5758.
Pascual G, Avgustinova A, Mejetta S, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541(7635):41–5.
Shan T, Zhang P, Jiang Q, **ong Y, Wang Y, Kuang S. Adipocyte-specific deletion of mTOR inhibits adipose tissue development and causes insulin resistance in mice. Diabetologia. 2016;59(9):1995–2004.
**ong Y, Xu Z, Wang Y, Kuang S, Shan T. Adipocyte-specific DKO of Lkb1 and mTOR protects mice against HFD-induced obesity, but results in insulin resistance. J Lipid Res. 2018;59(6):974–81.
Lee G, Zheng Y, Cho S, et al. Post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell. 2017;171(7):1545–58.
Vancura A, Nagar S, Kaur P, Bu P, Bhagwat M, Vancurova I. Reciprocal regulation of AMPK/SNF1 and protein acetylation. Int J Mol Sci. 2018;19(11):3314.
Xu YH, Song QQ, Li C, et al. Bouchardatine suppresses rectal cancer in mice by disrupting its metabolic pathways via activating the SIRT1-PGC-1α-UCP2 axis. Eur J Pharmacol. 2019;854:328–37.
Park EJ, Kim YM, Kim HJ, et al. (S)YS-51, a novel isoquinoline alkaloid, attenuates obesity-associated non-alcoholic fatty liver disease in mice by suppressing lipogenesis, inflammation and coagulation. Eur J Pharmacol. 2016;788:200–9.
Long JK, Dai W, Zheng YW, Zhao SP. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol Med. 2019;25(1):26.
Cheng J, Liu C, Hu K, et al. Ablation of systemic SIRT1 activity promotes nonalcoholic fatty liver disease by affecting liver-mesenteric adipose tissue fatty acid mobilization. Biochim Biophys Acta Mol Basis Dis. 2017;1863(11):2783–90.
MacNeil DJ, Howard AD, Guan X, et al. The role of melanocortins in body weight regulation: opportunities for the treatment of obesity. Eur J Pharmacol. 2002;450(1):93–109.
Damm E, Buech TR, Gudermann T, Breit A. Melanocortin-induced PKA activation inhibits AMPK activity via ERK-1/2 and LKB-1 in hypothalamic GT1-7 cells. Mol Endocrinol. 2012;26(4):643–54.
Lu J, Fang B, Huang Y, Tao S, Sun B, Guan S, ** Y. Epigallocatechin-3-gallate protects against 1,3-dichloro-2-propanol-induced lipid accumulation in C57BL/6J mice. Life Sci. 2018;209:324–31.
Wang X, Jiang H, Zhang N, Cai C, Li G, Hao J, Yu G. Anti-diabetic activities of agaropectin-derived oligosaccharides from Gloiopeltis furcata via regulation of mitochondrial function. Carbohydr Polym. 2020;229: 115482.
Kari S, Vasko VV, Priya S, Kirschner LS. PKA activates AMPK through LKB1 signaling in follicular thyroid cancer. Front Endocrinol (Lausanne). 2019;10:769.
Djouder N, Tuerk RD, Suter M, et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010;29(2):469–81.
Zahid H, Simpson ER, Brown KA. Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr Opin Pharmacol. 2016;31:90–6.
Brown KA, McInnes KJ, Hunger NI, Oakhill JS, Steinberg GR, Simpson ER. Subcellular localization of cyclic AMP-responsive element binding protein-regulated transcription coactivator 2 provides a link between obesity and breast cancer in postmenopausal women. Cancer Res. 2009;69(13):5392–9.
Swami S, Krishnan AV, Williams J, et al. Vitamin D mitigates the adverse effects of obesity on breast cancer in mice. Endocr Relat Cancer. 2016;23(4):251–64.
Yao Q, Li S, Cheng X, Zou Y, Shen Y, Zhang S. Yin Zhi Huang, a traditional Chinese herbal formula, ameliorates diet-induced obesity and hepatic steatosis by activating the AMPK/SREBP-1 and the AMPK/ACC/CPT1A pathways. Ann Transl Med. 2020;8(5):231.
Wang MD, Wu H, Fu GB, et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology. 2016;63(4):1272–86.
Indraccolo S, De Salvo GL, Verza M, et al. Phosphorylated acetyl-CoA carboxylase is associated with clinical benefit with regorafenib in relapsed glioblastoma: REGOMA trial biomarker analysis. Clin Cancer Res. 2020;26(17):4478–84.
Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. 2016;35(10):1250–60.
Lewis CA, Brault C, Peck B, et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. 2015;34(40):5128–40.
Giampietri C, Petrungaro S, Cordella M, et al. Lipid Storage and autophagy in melanoma cancer cells. Int J Mol Sci. 2017;18(6):1271.
Shi Y, Fan Y, Hu Y, et al. α-Mangostin suppresses the de novo lipogenesis and enhances the chemotherapeutic response to gemcitabine in gallbladder carcinoma cells via targeting the AMPK/SREBP1 cascades. J Cell Mol Med. 2020;24(1):760–71.
Ma X, Zhao T, Yan H, et al. Fatostatin reverses progesterone resistance by inhibiting the SREBP1-NF-κB pathway in endometrial carcinoma. Cell Death Dis. 2021;12(6):544.
Peck B, Schug ZT, Zhang Q, et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. 2016;4:6.
Huang J, Fan XX, He J, et al. SCD1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget. 2016;7(26):39970–9.
Wang J, Xu Y, Zhu L, et al. High expression of stearoyl-CoA desaturase 1 predicts poor prognosis in patients with clear-cell renal cell carcinoma. PLoS ONE. 2016;11(11): e0166231.
Han S, Wei R, Zhang X, et al. CPT1A/2-mediated FAO enhancement-a metabolic target in radioresistant breast cancer. Front Oncol. 2019;9:1201.
Pucci S, Zonetti MJ, Fisco T, et al. Carnitine palmitoyl transferase-1A (CPT1A): a new tumor specific target in human breast cancer. Oncotarget. 2016;7(15):19982–96.
Qian J, Chen Y, Meng T, et al. Molecular regulation of apoptotic machinery and lipid metabolism by mTORC1/mTORC2 dual inhibitors in preclinical models of HER2+/PIK3CAmut breast cancer. Oncotarget. 2016;7(41):67071–86.
Al-Zoughbi W, Pichler M, Gorkiewicz G, et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget. 2016;7(23):33832–40.
Wang YY, Attané C, Milhas D, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2017;2(4): e87489.
Balaban S, Shearer RF, Lee LS, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5:1.
Li N, Wang Y, Neri S, et al. Tankyrase disrupts metabolic homeostasis and promotes tumorigenesis by inhibiting LKB1-AMPK signalling. Nat Commun. 2019;10(1):4363.
Park YK, Obiang-Obounou BW, Lee KB, Choi JS, Jang BC. AZD1208, a pan-Pim kinase inhibitor, inhibits adipogenesis and induces lipolysis in 3T3-L1 adipocytes. J Cell Mol Med. 2018;22(4):2488–97.
Pierce SR, Fang Z, Yin Y, et al. Targeting dopamine receptor D2 as a novel therapeutic strategy in endometrial cancer. J Exp Clin Cancer Res. 2021;40(1):61.
Avtanski DB, Nagalingam A, Bonner MY, Arbiser JL, Saxena NK, Sharma D. Honokiol activates LKB1-miR-34a axis and antagonizes the oncogenic actions of leptin in breast cancer. Oncotarget. 2015;6(30):29947–62.
Watanabe S, Yaginuma R, Ikejima K, Miyazaki A. Liver diseases and metabolic syndrome. J Gastroenterol. 2008;43(7):509–18.
Li YC, Qiao JY, Wang BY, Bai M, Shen JD, Cheng YX. Paeoniflorin ameliorates fructose-induced insulin resistance and hepatic steatosis by activating LKB1/AMPK and AKT pathways. Nutrients. 2018;10(8):1024.
Gu L, Cai N, Lyu Y, et al. γ-Mangostin ameliorates free fatty acid-induced lipid accumulation via the SIRT1/LKB1/AMPK pathway in HepG2 and L02 cells. J Agric Food Chem. 2019;67(50):13929–38.
Li X, Zhang Y, ** Q, et al. Liver kinase B1/AMP-activated protein kinase-mediated regulation by gentiopicroside ameliorates P2X7 receptor-dependent alcoholic hepatosteatosis. Br J Pharmacol. 2018;175(9):1451–70.
Zeng Y, Hua YQ, Wang W, Zhang H, Xu XL. Modulation of SIRT1-mediated signaling cascades in the liver contributes to the amelioration of nonalcoholic steatohepatitis in high fat fed middle-aged LDL receptor knockout mice by dihydromyricetin. Biochem Pharmacol. 2020;175: 113927.
Wang Z, Yang X, Kai J, et al. HIF-1α-upregulated lncRNA-H19 regulates lipid droplet metabolism through the AMPKα pathway in hepatic stellate cells. Life Sci. 2020;255: 117818.
Lin L, Zeng L, Liu A, et al. l-Theanine regulates glucose, lipid, and protein metabolism via insulin and AMP-activated protein kinase signaling pathways. Food Funct. 2020;11(2):1798–809.
Svensson RU, Parker SJ, Eichner LJ, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22(10):1108–19.
Siqingaowa SS, Gopalakrishnan V, Taghibiglou C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem Biophys Res Commun. 2017;488(1):136–40.
Liu G, Kuang S, Cao R, Wang J, Peng Q, Sun C. Sorafenib kills liver cancer cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids via the ATP-AMPK-mTOR-SREBP1 signaling pathway. FASEB J. 2019;33(9):10089–103.
Gugiatti E, Tenca C, Ravera S, et al. A reversible carnitine palmitoyltransferase (CPT1) inhibitor offsets the proliferation of chronic lymphocytic leukemia cells. Haematologica. 2018;103(11):e531–6.
Luo J, Hong Y, Lu Y, et al. Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the warburg effect by cetuximab. Cancer Lett. 2017;384:39–49.
Talebi A, Dehairs J, Rambow F, et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat Commun. 2018;9(1):2500.
Funding
This work was supported by International Cooperation Project of the Department of Science and Technology of Jilin Province (20190701006GH); National Natural Science Foundation of China (8201001069); and Jilin Province Health and Health Technology Innovation Project (2020J033).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Ethical approval
The manuscript does not contain any studies with human participants or animals.
Informed consent
Informed consent statements are not applicable for this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Geng, J., Zhang, Y., Meng, Q. et al. The role of liver kinase B1 in tumor progression through regulation of lipid metabolism. Clin Transl Oncol 24, 2045–2054 (2022). https://doi.org/10.1007/s12094-022-02863-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-022-02863-2